|
|
||||||
|
|||||||
|
|||||||
|
|||
|
|||
Molecular Mechanisms of the Transition from Myocardial Hypertrophy to FailureKenneth B. MarguliesUniversity of Pennsylvania, Philadelphia, USAMargulies reviewed recent data that support the hypothesis that adaptive signaling promotes hypertrophy with improved or preserved function and thus suppresses the stimulus and hence physiological hypertrophy does not progress to heart failure. He also reviewed data showing some determinants of the progression from pathological hypertrophy in some situations.
No progression from physiological hypertrophy to failure: Why? Little overlap in the patterns of altered gene expression observed after aortic banding (pathological stimulus) and prolonged swimming (physiological stimulus) was demonstrated by Izumo and Mann.
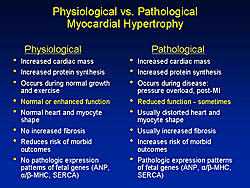
The major differences between physiologic and pathological hypertrophy are shown in Figure 1. The pivotal difference, Margulies stated, is that physiological hypertrophy is consistently associated with preserved or even enhanced myocardial contractile function, while pathological hypertrophy is sometimes associated with reduced contractile function, a circumstance that triggers various local and systemic adaptations and additional pathological signaling. Recent work by DeBosch, Muslin and colleagues demonstrated the central importance of Akt-dependent signaling as a pivotal node in distinguishing physiological and pathological cardiac growth. In Akt knockout mice, they showed the animals had a normal phenotype and lifespan with normal echocardiographic dimensions and function. But, stimulation with physiological growth factors, such as IGF and growth hormone, reduced cardiac protein synthesis. In response to prolonged swimming, the wild type mice showed increased cardiac size and myocyte cross-sectional area, while the heterozygous and homozygous Akt knockouts had no changes in cardiac and myocyte growth. Interestingly, in response to pathological stress, the Akt knockout mice had increased in vitro protein synthesis with endothelin simulation, which is known to trigger pathologic hypertrophic signaling, and reduced cardiac function in vivo. Significantly increased pathological hypertrophy with aortic banding also occurred, with an increase in the size of the heart and myocytes, and increased cardiac ANF expression in the homozygous knockout mice. Their work strongly suggests that Akt-dependent signaling is involved with physiological hypertrophy and that Akt signaling also exerts a restraining effect on the hypertrophy associated with pathological stimuli like sustained pressure overload.
Determinants of progression from pathological hypertrophy to heart failure Margulies reviewed the hypothesis that depressed contractile function with defective calcium (Ca) homeostasis triggers Ca-dependent signaling (calcineurin, PKC and/or CaMK) that drives pathologic transcription and exacerbates dysfunction. Studies of excitation-contraction coupling and basic Ca cycling dynamics in hypertrophied and failing hearts have shown that a positive force frequency response indicates relatively intact calcium cycling dynamics and a negative or flat force-frequency response indicates impaired EC coupling and defects in myocyte calcium cycling. Work from Margulies’ laboratory in human myocardium showed that in nonfailing muscles there is a clear positive force-frequency response, while failing muscles display a sharp negative force-frequency response. They also found elevated tension developed in failing muscles at subphysiological pacing rates, suggesting an inability of the myocyte to rid itself of calcium when appropriate. These failure-associated defects in Ca cycling dynamics have often been associated with alterations in the abundance of Ca handling proteins such as reduced abundance of the sarcoplasmic reticulum ATPase (SERCA), increases in the sodium calcium exchanger (NCX), or both. Chen, Houser and colleagues showed in lab-generated mice with inducible overexpression of the Beta-2a subunit of the L-type Ca channel increases in the L-type Ca channel current, Ca2+ transients, contractions, and the Na/Ca exchanger current density relatively early after activation of the inducible transgene. These mice simultaneously developed decreases in SERCA abundance and increases in NCX abundance. Further, gene-dose-related cardiac hypertrophy and myocardial fibrosis with reduced in vivo fractional shortening was found among the higher expressing mice. Interestingly, they showed that metoprolol, a Beta 1-adrenergic blocker, reduced the damaging effects of Cav1.2b2a expression in the high-expressing line. Metoprolol reduced cardiac hypertrophy, normalized fractional shortening, and diminished fibrosis in the high-expressing line. Conversely, an ISO concentration that only caused a modest injury in control (and Cav1.2b2a on DOX) caused rapid death in high expressing Beta-2a mice. In low expressing mice, ISO also increased death rates, decreased cardiac function, and enhanced myocyte loss and fibrosis. Together, these data show that a molecular manipulation that initially induced an isolated increase in myocyte calcium entry with enhanced contractile function, subsequently triggered molecular adaptations, including decreased SERCA and increased sodium-calcium exchanger. These effects resulted in reduced contractility, hypertrophy with increased myocyte loss, and increased fibrosis. Further, this resulted in a particular sensitivity to catecholamines and a protective effect by beta-blockers. These are all effects that are characteristic of the transition from hypertrophy to failure in human hearts, explained Margulies. A study by Wu and colleagues demonstrated that G-protein coupled receptor (GPCR binding triggers inositol tri-Phosphate (IP3) generation, then IP3 binds to IP3 receptors on the nuclear envelope and induces increased calcium within an intranuclear microdomain. This localized calcium increase activates CaMKII and PKD to induce nuclear export of HDAC5 and transcription. Notably, the calcium concentration at the nuclear envelope was linked to the calcium load in the sarcoplasmic reticulum, while that in the microdomain was highly buffered to allow controllable calcium-dependent HDAC export and transcription in the midst of cyclic micromolar changes in overall cytoplasmic calcium. Further work by Wu and colleagues in this study showed with endothelin stimulation a time-dependent HDAC export from the nucleus. This nuclear export was not altered by PKC inhibition, but is dependent on IP3 receptor binding. Further, this signaling pathway involved in excitation-transcription coupling was abolished in an IP3 receptor knockout mouse. The calcineurin-NFAT pathway has also been implicated in the transition from hypertrophy to failure. Liang and Molkentin have proposed that calcineurin, a calcium-sensitive phosphatase, triggers dephosphorylation of NFAT resulting in NFAT translocation into the nucleus and activation of pathological transcription. The factors that favor NFAT dephosphorylation also favor represssion of pathological transcription. Molkentin and colleagues, using a transgenic mouse model with an NFAT-luciferase reporter, demonstrated that increased NFAT activity clearly preceded increased hypertrophy in the setting of aortic-banding pressure overload and that particularly pronounced NFAT activity was present late after banding when decompensation was typically observed. Further, NFAT activity was higher in mice with experimental myocardial infarctions, increased lung weight, and reduced cardiac function, compared to mice with compensated hypertrophy with lesser dysfunction and no increases in lung water. Therefore, graded activation of calcineurin NFAT signaling is clearly implicated in the transition from compensated hypertrophy to decompensated failure Notably, decreases in NFAT activity, even as cardiac mass was increasing, were shown in the studies by Wilkins and colleagues where physiological hypertrophy was induced by prolonged swimming. Combined with the results in the Akt knockout mice, these results clearly demonstrate distinct and reciprocally regulated signaling with physiological and pathological hypertrophy.
Summary Physiological hypertrophy, from exercise or normal somatic growth, involves endogenous growth factors like GH and IGF that trigger an Akt-dependent transcriptional program that produces hypertrophy with normal or enhanced function. This normal or enhanced cardiac function allows the heart to meet the increased demand without support by compensatory mechanisms like catecholamines, ET1 or other detrimental GPCR agonists and allows a negative feedback response. In contrast, pathological stress, from MI or overload, triggers clearly distinct signaling pathways, including Ca-dependent signaling like CaM Kinase and Calcineurin/NFAT pathways, and activates a pathological transcriptional pathway that is often associated with reduced or depressed myocardial function. When function is sufficiently depressed, compensatory activation of catecholamines, angiotensin II and endothelin occurs and triggers even more pathological Ca-dependent signaling with exacerbation of hypertrophy and dysfunction. Thus, particularly when pathological stimuli are associated with a critical level of myocardial dysfunction, a positive feedback loop is created and a transition to decompensation and failure is far more likely.Therefore, blocking persistent stimuli and antagonizing compensatory neurohumoral activation are two possible ways to block the progression from pathological hypertrophy to failure. Future therapeutic strategies may include those that support improved function without agonism of GPCR, that directly block Ca2+-dependent signaling, and promote activation of physiological signaling pathways. |
Role of Ryanodine Receptor in Heart FailureMasofumi YanoYamaguchi University School of Medicine, Yamaguchi, JapanAbnormal regulation of intracellular calcium by the sarcoplasmic reticulum (SR) is the chief pathogenic mechanism for various types of dysfunctions in heart failure. Dr. Masofumi Yano reviewed a new strategy for the treatment of heart failure by stabilizing the calcium release channel of cardiac sarcoplasmic reticulum, often referred to as the ryanodine receptor (RyR). In the setting of heart failure, three or four FKBP become dissociated from the RyR with compensatory changes to the RyR that induce abnormal calcium leak, as reported by Yano and colleagues in Circulation in 2000. Marx and colleagues reported that the cause of the dissociated FKBP is protein kinase A (PKA) phosphorylation. The diastolic calcium leak is recognized as increased calcium sensitivity at the single channel level. In the normal channel, there is a linear increase in calcium concentration and open channel variability. Importantly, the channel is usually closed when the calcium concentration is below the level of 0.1 µM, which corresponds with the diastolic calcium concentration. However, in the failing heart, the channel opens when the calcium level is decreased during diastole. This is considered the basic characteristic of the abnormal calcium leak. In malignant hyperthermia, a single-point mutation disease of the skeletal RyR, this abnormal channel gating is seen, suggesting a common abnormality within the RyR is involved in heart failure and malignant hyperthermia. More than 80 mutations have been found in malignant hyperthermia and central core disease (CCD), centered around three major regions, the N-terminal domain, the central domain, and the channel-forming C-terminal region. More than 40 mutations of the RyR have been reported in patients with arrhythmogenic right ventricular cardiomyopathy (ARVC) or catecholaminergic polymorphic ventricular tachycardia (CPVT). Interestingly, the FKBP binding site is closely located to the central mutable domain, suggesting that a single-point mutation and FKBP dissociation may share a common conformational change leading to the defective channel gating. An intriguing hypothesis was proposed by Ikemoto and colleagues that the N-terminal domain and central domain interact with each other as a regulatory switch for the channel gating. Yano and colleagues performed work to determine if this hypothesis could be applied to the RyR. In the normal channel, the regulatory domains interact with each other in the zipped state, but if there is a mutation at either the N-terminal or central domain, domain unzipping occurs and this could induce calcium leak; this situation was mimicked using the domain peptide approach by Yano and colleagues. Work strongly supporting the domain switch hypothesis was reported by Liu and colleagues showing the N-terminus and central domain are closely interacting and very closely located. Yano and colleagues quantitated the state of domain unzipping by chemical quencher experiments and showed that the domain peptide DPc10 induces domain unzipping. Interestingly, FK506, which causes dissociation of FKBP, also induces domain unzipping. Further, they showed that a calcium leak was induced by DPc10 in a dose-dependent manner. In the failing heart, they showed that the SR is PKA-hyperphosphorylated and the FKBP is dissociated from the RyR; spontaneous calcium leak was observed. Interestingly, in such failing SR, the domain unzipping already occurred to the level seen in the DPc10-induced normal SR. Together these data show that in the normal condition, the channel regulation is in the zipped state and the channel is stabilized. But, an inherent single point mutation or FKBP dissociation from the Ryr receptor induces domain unzipping and leads to abnormal calcium leak. Yano and colleagues searched for a reagent that reverses the mode of domain unzipping to zipping to inhibit the abnormal calcium leak. They hypothesized that if there is a common abnormality within the cardiac RyR that dantrolene would be effective to restore defective channel gating, based on previous work with this reagent. They showed that dantrolene completely corrected the defective interdomain interaction, that is, shifted the mode from zipping to unzipping, and thereby prevented calcium leak. In summary, excess beta-stimulation induces PKA-mediated hyperphosphorylation of the RyR, which leads to dissociation of FKBP from RyR, and thus leads to domain unzipping of the regulatory domain that results in calcium leak. These events lead to intracellular calcium overload, cardiac dysfunction, heart failure, and arrhythmia. A single point mutation seen in ARVC directly induces the domain unzipping of the regulatory domain. The defective domain interaction plays a critical role in the abnormal calcium handling in heart failure, and correction of this defect would be a potent new therapeutic strategy against heart failure. |
|
|
|
Copyright © 2007 Japanese Circulation Society All Rights Reserved. webmaster@j-circ.or.jp |