|
|
||||||
|
|||||||
|
|||||||
|
|||
|
|||
|
|
|
Oxidant stress is defined as excess production of reactive oxygen species (ROS) that outstrips antioxidant defenses. Oxidant stress is implicated in pathobiological processes in which oxidation of biological macromolecules, such as protein, DNA, and lipids, occurs, and it is involved in the pathogenesis of many cardiovascular diseases. Oxidant stress is evoked by risk factors for coronary heart disease. The cell can attempt to modulate oxidant stress that accompanies normal metabolism in a variety of ways. Low molecular weight antioxidants, such as vitamin C and D, and a host of enzymatic antioxidants are important in minimizing the flux of reactive oxygen species and their toxicity. Glutathione peroxidases and glucose-6 phosphate dehydrogenase (G6PD), two antioxidants, were the focus of this lecture by Dr. Joseph Loscalzo of Brigham and Women’s Hospital, Boston, USA. Glutathione peroxidases and G6PD combat the oxidant stress caused by agents like superoxide peroxides, peroxynitrite, and nitric oxide. The reactive oxygen species are related through a variety of very rapid chemical reactions and intermediates. Superoxide represents the first of the partially reduced forms of molecular oxygen, which is then converted through dysmutation to hydrogen peroxide which is then reduced to water. The dismutation of superoxide to hydrogen peroxide can occur non-enzymatically but the superoxide dismutases, of which there are three, will catalyze this reaction approximately eight-fold. Hydrogen peroxide is converted to water and lipid peroxides to lipid alcohols by the action of glutathione peroxidase, which requires glutathione as a cosubstrate. Catalase will very rapidly convert hydrogen peroxide to water, but not lipid peroxides to lipid alcohols. Importantly even though it is very fast at this conversion, it has a reasonably weak KM for the substrate so that lower concentrations of hydrogen peroxide typically found under normal cellular metabolism conditions need to be eliminated by the glutathione peroxidases. In the absence of adequate enzymatic catalysts, these ROS species will react through well-known chemical mechanisms like Haber-Weiss chemistry or Fenton chemistry, which uses reduced transition metals to produce very highly reactive species that are very damaging to tissues; hydroxyl radical and hydroxide anion.
In the central antioxidant enzyme cascade is initiated by the reduction of peroxides to alcohols by the glutathione peroxidases. This requires glutathione which is converted from the disulfide by the action of glutathione reductase, which in turn requires NADPH as an obligate reducing substrate. The principal source of any DPH in the cytosol is glucose-6 phosphate dehydrogenase as the rate-limiting enzyme for the synthesis of pentose phosphates. However, the most crucial role of G6PD is the synthesis of any DPH. Three oxidative enzymopathies were discussed by Loscalzo: Glutathione peroxidase-1 deficiency, glucose-6-phosphate dehydrogenase deficiency, and glutathione peroxidase-3 deficiency (Figure 1).
Homocysteine theory of atherothrombosis The relationship of homocysteine to atherothrombosis was first proposed by McCully in 1969 from his study of autopsy materials of young individuals who died with the rare in-born error of metabolism homocysteineuria, where homocysteine levels are very high. These patients had arteriosclerosis with fibrosis and proliferative lesions. He hypothesized that mild forms of hyperhomocysteinemia could contribute to atherosclerotic risk. The epidemiologic relationship between mild to moderate elevations of homocysteine and the risk of atherothrombotic events is supported by evidence from more than 40 studies. Mild hyperhomocysteinemia (11 to 20 micromolar) is found in 20-40% of patients with vascular disease, but in only 2% of unaffected individuals. A meta-analysis by Boushey showed that mild hyperhomocysteinemia is associated with a significant increase in the relative risk of events in the coronary, cerebral, and peripheral arterial circulation. A dose-dependent relationship was found between homocysteine levels post-myocardial infarction (MI) and subsequent events, with the highest homocysteine levels associated with worse proportional survival, in a study in Scandinavia published by Nygard in 1997.
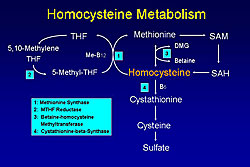
Homocysteine can be eliminated by trans-sulphuration, which occurs only in the liver and spleen, or by remethylation, which can occur in all cells, particularly vascular cells as the only mechanism for lowering homocysteine in those cells (Figure 2). The metabolism of homocysteine requires B vitamins, especially folate, B12, and B6. Homocysteine is part of the remethylation cycle, thus its level regulates the ratio of S-adenocyle methionine (SAM), which is the only methyl donor in cells for all methylation reactions. Homocysteine regulates the ratio of SAM to S-adenocyle homocysteine (SAH), and this ratio sets the propensity to methylamine reactions in cells. A variety of genetically-acquired enzyme deficiencies or abnormalities have been associated with hyperhomocysteinemia, including cystathionine-beta synthase (CBS), the rate-limiting enzyme in the trans-sulphuration pathway. To determine if mild hyperhomocysteinemia can cause atherothrombosis, Loscalzo and colleagues used a CBS-deficient mouse model to assess endothelial function as an early marker of a propensity to vascular disease. Homocysteine levels in rodents are about one-half that seen in humans. Nitric oxide (NO) and its bioactivity were used as one measure of normal endothelial function. A heterozygous-deficient state of CBS was accompanied by a paradoxical vasoconstrictor response to methacholine, which normally would cause vasodilation in the wild type animals. These animals were studied at 16 to 20 weeks of age. Their vessels looked morphologically normal and they responded normally to nitroprusside. Upchurch and colleagues showed that homocysteine caused a dose-dependent reduction in NO production in cultured endothelial cells. Similarly, the vasodilator response and cyclic GMP (cGMP) production were reduced in aortic rings from heterozygous-deficient CSB animals. These vascular rings comprised about one-half the normal cGMP in response to an agonist that stimulated NO release. Thus, these data suggest there is less bioactive NO produced by the endothelial cells exposed to elevated homocystine levels. A reduction in glutathione peroxidase-1 (GPx-1), a key antioxidant enzyme found in all mammal cell types, was caused by hyperhomocysteinemia in the experiments by Loscalzo and colleagues. No effect on the role of nitric oxide synthase (NOS) expression or activity was found. A reduction in glutathione peroxidase-1 activity would lead to an increase in H2O2 and LOOH peroxides, derivative of which would react with and inactivate NO. Handy and colleagues increased the intracellular synthesis of homocysteine, rather than adding it exogenously, by boosting selenium concentrations and subsequently inhibited remethylation with aminopterin and methionine. This resulted in a 2-fold increase in homocysteine in the cell and a halving of GPx-1 expression. How does homocysteine suppress GPx-1 expression? Selenocysteine, the twenty-first amino acid, contains a selenium atom in place of sulphur. The synthesis of selenocysteine is unusual because its codon is UGA, which is the stop codon. Crucial translational cofactors, including SBP2, eEFsec, DNA binding protein B, and nucleolin allow the ribosome to read UGA as coding for selenocysteine-charged TRNA. Work by Handy showed that if the codon was read through PGA or UGA as coding for selonocysteine it would yield mature luciferase that luminesced. If the UGA codon were read as stock, there would be no luminescence. This elegant assay for monitoring translation showed that homocysteine in the presence of adenosine and especially adenosine deaminase inhibitor would suppress the expression of mature luciferase. An effect by homocysteine on methylation is suggested by these data, because adenosine was required for an optimal effect. One piece of clinical evidence supports the view that mild hyperhomocysteinemia suppresses methylation. Ingrosso and colleagues showed that subjects with the highest levels of homocysteine have lower levels of DNA methylation. Global methylation is attenuated by hyperhomocysteinemia, although the targets for the methylation change are not unknown Loscalzo and colleagues showed that CBS heterozygous-deficient animals made abundant superoxide compared to wild-type animals and that this is accompanied by an increase in 3- nitrotyrosine (3-NT) in aortic tissue, a footprint of inactivated oxidized nitric oxide in tissue. Further, GPx1-deficient animals they showed an increase in 3-NT consistent with the hypothesis. They were able to restore the normal phenotype by treating the CBS heterozygous animals with pharmacologic therapy to restore cysteine and glutathione levels or crossing the CBS heterozygous animals with animals that had a GPx-1 transgene that allowed for about a 2- to 3-fold increase in GPx-1 expression. The paradoxical vasoconstrictor response was restored to the normal response when the animals were given a glutathione-sustained precursor. Crossing the GPx-1 overexpressers into the CBS-heterozygous background animals restored the normal vasodilator response compared to the CBS response alone. These data are fairly consistent with the hypothesis that hyperhomocysteinemia causes an oxidative enzymopathy that is accounted for by decreased translation of GPx-1. Epidemiologic data consistent with the importance of GPx-1 comes from work by Schnabel and colleagues and published in 2005. In a large population of patients with atherothrombotic disease, a homoscysteine level above the median and a GPx-1 level below the median were associated with worse event-free survival. An elevated GPx-1 level was a negative risk factor for events. Together, these data reviewed by Loscalzo show that an elevated homocysteine level causes an oxidative enzymopathy through its induction of an acquired form of GPx-1 deficiency.
G6PD deficiency and its relationship to oxidant stress in the vasculature The G6PD deficiency is the most common enzymopathy worldwide, with about 400 million cases. There are over 400 different biochemical variants and over 120 different mutations, all of which are single-point mutations or small deletions. Eliminating the gene leads to embryonic lethality. Leopold in Loscalzo’s group hypothesized that G6PD deficiency promotes oxidant stress and vascular dysfunction. The key role of G6PD is generation of NADPH, essential for generating glutathione. Glutathione is not only a cofactor for glutathione peroxidase reactions, it is also important for optimal activity of endothelial NOS. NADPH is also a cofactor for the synthesis of tetrahydrobiopterin, also a key cofactor for endothelial NOS. Thus, from an NADPH-centric view of the cell, any effect on its reduction should have drastic consequences for endothelial function and NO production. Leopold’s work has shown that suppressing G6PD in endothelial cells with antisense or sRNA increased oxidant stress and that overexpressing G6PD with an adenoviral factor decreased oxidant stress. Further, a concomitant decrease in NADPH with suppression and increase in NADPH with overexpression occurred. Changes in NO production also occurred. Suppressing G6PD reduced measurable NO levels and overexpressing G6PD increased NO levels dramatically. Interestingly, eNOS and G6PD would coprecipitate in immunoprecipitation immunoblotting experiments done in either order. Changes in NO bioactivity were also found. Suppressing G6PD expression with dehydroepiandrosterone (DHEA) resulted in a significant reduction in cGMP production in response to typical endothelial agonists. Regarding the role of G6PD in angiogenesis, Leopold showed that suppressing G6PD with antisense resulted in a reduction in endothelial tube formation or overexpressing it with a virus resulted in an increase in tube formation. In an animal model of G6PD deficiency, males had a significant attenuation of neovascular outgrowth in organ culture assays when compared to the wild-type animals in response to vascular endothelial growth factor (VEGF). There were correlative changes in phosphotyrosine receptor events in endothelial cells. There were correlative changes in phospho-AKP generation and in phospho-eNOS generation, which decreased with suppression and increased with overexpression. In isolated hearts, G6PD deficiency had a clear effect on ventricular function. Work done with Jain and colleagues showed there was in an increase in end-diastolic pressure in G6PD-deficient animals and a decrease in developed pressure when compared to the wild types in response to no-flow ischemia. In isolated cardiomyocytes, there was an increase in oxidant stress in the G6PD-deficient animals when compared to the wild types. Forgione and colleagues showed in African American men that G6PD deficiency was accompanied by a significant reduction in flow-mediated dilator response and nitroglycerin-dependent dilation. These changes were associated with an increase in markers of oxidant stress. About 15% of the total African American population has G6PD deficiency.
Acquired G6PD deficiency from mild hyperaldosteronemia Aldosterone has known adverse effects on endothelial function, vascular compliance, tissue injury, and ischemia. There is also reasonable evidence showing aldosterone has effects on reactive oxygen species generation and nitric oxide bioactivity reduction. Loscalzo and colleagues examined whether aldosterone has an effect on G6PD activity because of its structural relationship to DHEA, the only known pharmacologic agent to be a reasonably selective inhibitor of G6PD. In subjects undergoing normal vascular function studies, they found a very good negative correlation between G6PD activity and aldosterone, over a range of aldosterone that would otherwise be considered normal. Leopold then showed that aldosterone reduces G6PD expression, mRNA expression, and NADPH, showing that aldosterone causes an acquired form of G6PD deficiency. Also, eNOS activity is decreased without a change in expression and cGMP levels decrease as a function of increasing aldosterone concentrations. This effect appears to be mediated by the mineralocorticoid receptor because spironolactone can reverse its effects. This was shown for NADPH levels, DCF fluorescence, and eNOS activity. The mineralocorticoid receptor appears to have its effect by acting through protein kinase A (PKA). PKA activity was shown to increase with aldosterone, without a change in expression, accompanied by an increase in phospho-CREB and an increase in CREM. Further work by Leopold led to a working hypothesis that aldosterone, acting through the mineralocorticoid receptor and PKA, increases phosphorylation of CREB, but more potently increases CREM, which suppresses pre-dependent expression of G6PD. This in turn leads to a decrease in NADPH, GSH, GPx-1 activity, uncoupling of eNOS, and a decrease in bioactive NO. Testing this hypothesis in an animal model infused with aldosterone via a minipump showed that significant increases in aldosterone levels were accompanied by a decrease in G6PD activity, an increase in superoxide production, a decrease in aortic cGMP production, and a decrease in NADPH in aortic tissue. Further, overexpressing G6PD in the same animals, restored normal endothelial function and significantly improved superoxide production. These data suggest there are two different mechanisms by which G6PD can lead to oxidant stress. One is inherited and one is acquired by mild hyperaldosteronemia.
Glutathione peroxidase-3 deficiency Two brothers presented with vascular events. The 7-year-old boy had 2 cardiovascular accidents (CVA) and the 4-year-old boy had 2 transischemic attacks (TIAs). The second event in each child occurred while there on aspirin therapy. Initial routine screening for known prothrombotic states was negative. Remarkable insensitivity by their platelets to NO was found, which appeared to be due to an inactivation of NO by something in their plasma. Work by Loscalzo and colleagues showed the children had a deficiency of the extracellular isoform of glutathione peroxidase-3 (GPx-3), which conducts the same chemistry as GPx-1. It is made largely by renal tubular cells and it is the most important peroxidase in the extracellular compartment. GPx-3 is synthesized in the same way as GPx-1 and it is a selenocysteine-containing protein. The genomic structure of GPx-3 is more complicated, but the principles are the same. Both boys have less than half of the plasma activity and GPx-3 levels compared to age-matched controls and an anephric child. In these children, inhibition of platelet function could be restored by adding glutathione peroxidase to their plasma. The mother had an intermediate phenotype in these assays, but she had no evidence for a clinically important thrombotic event that she, as a nurse, recognized. The father and the unaffected sister were normal. Kenet and colleagues in Israel followed 12 families, 7 of whom had GPx-3 deficiency. An inverse relation between plasma GPx-3 activity and suppression of P-Selectin expression by NO donor. Voetsch and colleagues showed that a unique, less frequent haplotype (haplotype H2) was an independent risk for CVA in a study of 114 young CVA patients and 122 age-matched controls. The odds ratio for an CVA even in this large, unrelated population was 2.1. Environmental factors, especially smoking, and known genetic factors, including the eNOS polymorphism and the paraoxonase polymorphism, increased the risk. These data suggest that a deficiency of GPx-3 leads to an increase in the steady state levels of peroxides, which are normally released from activating platelets, leading to a reduction in bioactive NO available to suppress further platelet recruitment. This leads to an increase in the likelihood of platelet-dependent thrombosis in the cerebral vascular circulation. |
||||||

|
|
|
Copyright © 2007 Japanese Circulation Society All Rights Reserved. webmaster@j-circ.or.jp |