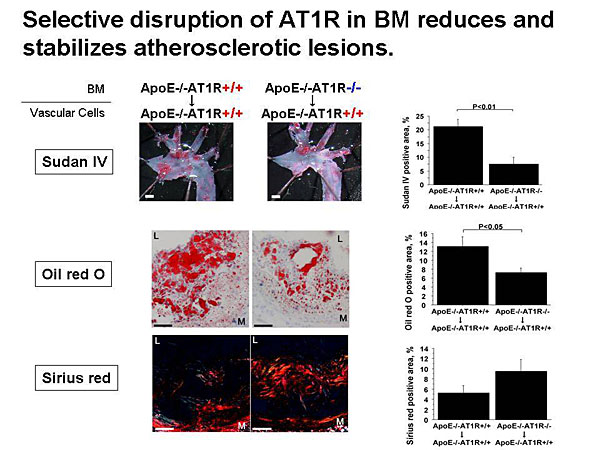
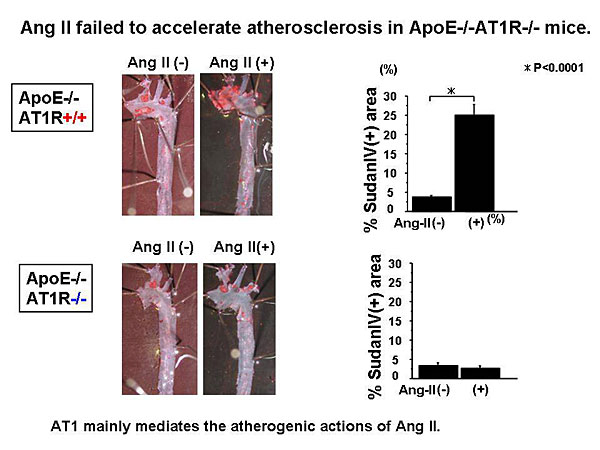
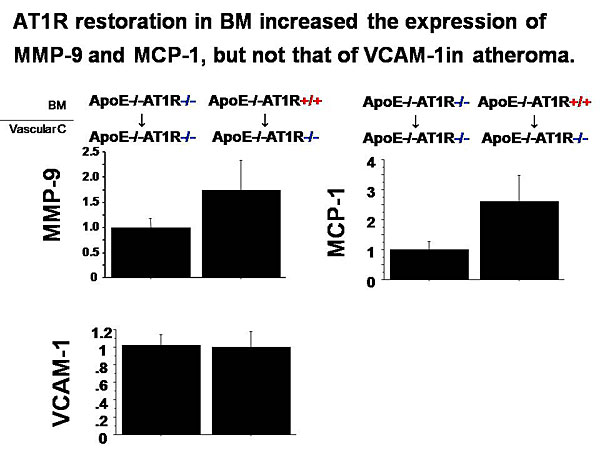
Dr. Masanori Aikawa, Brigham and Women’s Hospital, Harvard Medical School, presented data on the role of macrophages in atherogenesis and thrombosis and the effects of lipid-lowering on plaque inflammation and thrombotic complications.
Macrophages are thought to induce thrombotic complications of atherosclerosis through the action of matrix metalloproteinase (MMP) collagenases. Studies in collagenase-resistant and collagenase-deficient mutant mice demonstrated that MMP-collagenases regulate collagen accumulation and organization in mouse atheromata. These studies suggest that MMP-collagenases cause collagen loss in the plaques, leading to plaque vulnerability with subsequent rupture and thrombosis.
Preclinical and clinical studies of lipid-lowering therapy provide further support for the role of inflammation in thrombotic complications of atherosclerosis. Several large statin trials demonstrated a 15% to 40% risk reduction in acute atherosclerotic thrombotic complications without substantially reducing plaque burden.
From 1998 to 2006, Dr. Aikawa and colleagues tested the effects of lipid-lowering on atherosclerotic plaques in rabbit models. The studies demonstrated that lipid-lowering improved a number of features associated with plaque vulnerability and thrombogenicity. Tissue sections of atheromata from cholesterol-fed rabbits showed decreased macrophages after dietary lipid-lowering compared to baseline. Additional histologic studies revealed reduced MMP-collagenase expression and increased collagen accumulation and reduced expression and in situ binding of tissue factor in rabbit atheromata, suggesting potential mechanisms for plaque stabilization. Oxidized LDL (OxLDL) accumulation and endothelial cell activation were decreased from baseline suggesting a potential mechanism for reduced inflammation by lipid-lowering therapy.
A recent study showed that dietary lipid-lowering decreased oxidized phospholipids (OxPL) in rabbit atheromata but increased plasma OxPL in the same rabbits. The mechanisms for this are unclear but OxPL in the blood may serve as a biomarker for plaque stabilization or regression during anti-inflammatory therapy.
Current studies are focusing on molecular imaging of plaque macrophages. One study used 3.0 tesla MRI enhanced with magnetic iron nanoparticles to quantify macrophages in rabbit atheromata. Using this technique, Dr. Aikawa found that cholesterol lowering with statin therapy reduces the magnitude of signal loss in rabbit atheromata. In another study, intravital fluorescence microscopy showed that macrophage accumulation precedes osteogenesis in mouse plaques, suggesting that plaque calcification is related to inflammation. When the mice were treated with statins, macrophage accumulation and osteogenic activity both decreased.
Dr. Aikawa concluded that cholesterol lowering reduces inflammation in atherosclerosis and improves features associated with unstable plaques. Molecular imaging can monitor effects mediated by anti-inflammatory therapies such as lipid-lowering. |