|
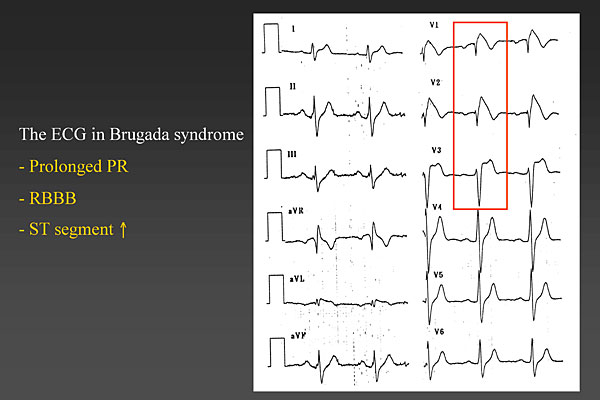
Figure 1. The ECG in Brugada syndrome.
【Click to enlarge】 |
|
|
Dr. Josep Brugada, Hospital Clinic, University of Barcelona, Spain, presented an update on the features, diagnosis, and treatment of Brugada syndrome. Brugada syndrome is a clinical-electrocardiographic diagnosis based on the occurrence of syncopal episodes or sudden death in patients without demonstrable structural heart disease. The landmark finding is a characteristic ECG pattern of apparent right bundle branch block (RBBB) and ST segment elevation in leads V1 to V3 (Figure 1). Most people with Brugada syndrome also have a prolonged PR interval. Sudden cardiac death in Brugada syndrome is caused by polymorphic ventricular tachycardia (VT) or ventricular fibrillation (VF).
The first patient with Brugada syndrome was identified in 1986, with the first published abstract on the syndrome appearing in 1991 and the first paper in 1992. Since then it has been learned that Brugada syndrome accounts for 1% to 5% of unexpected sudden deaths and up to 50% of sudden deaths in patients with a structurally normal heart. Brugada syndrome is one of the leading causes of natural death in males younger than 50 years in south Asia.
Several years ago a consensus document described three types of Brugada syndrome, based on the ECG pattern—Type 1 (coved) and Types 2 & 3 (saddle back). This was a mistake, cautioned Dr. Brugada, because Types 2 & 3 only indicate a suspicious ECG and are not Brugada syndrome. Only the Type 1 coved ECG identifies a patient with Brugada syndrome. Many conditions can mimic Brugada syndrome with an ST segment elevation, including a mediastinal tumor compressing the right ventricular outflow tract (RVOT), cocaine intoxication, acute myocardial infarction (MI), dissecting aortic aneurysm, and various central and autonomic system disorders, among many others. Additionally, the ECG of a patient with Brugada syndrome can vary considerably over time, appearing with the classic Brugada syndrome pattern at one time and in another instance appearing completely normal. Some patients might have a Brugada syndrome ECG while febrile but a normal ECG when not febrile.
Work done in Japan has produced methods using certain drugs to “unmask” or modulate the ECG. One drug, ajmaline reveals the ST elevation, which normalizes after isoproterenol administration. In a patient with isolated RBBB, administration of ajmaline may prolong the PR interval but it does not elevate the ST segment. The same is true for patients with arrhythmogenic right ventricular dysplasia (ARVD). The ajmaline test has been useful for identifying affected family members.
The sensitivity and specificity of the ajmaline test have been tested in families in whom the sodium channel gene (T1620M) associated with Brugada syndrome was identified. In one such analysis, of 147 family members, 104 were at risk and 61 were genotype positive, with 20 having a positive basal ECG and 41 having a negative basal ECG. Of the 41 with negative basal ECG, the ajmaline ECG was positive in 28, false-negative in 7, and not done in 6. Of the 43 genotype negative patients, none had a basal abnormal ECG, and the ajmaline ECG was false-positive in 2, negative in 34, and not done in 7. Dr. Brugada is suspicious that the 2 false-positive patients (2 sisters) might have a different mutation. From these studies, the sensitivity of the ajmaline test was calculated as 80%, specificity as 94%, positive predictive value (PV) as 93%, and negative PV as 83%. Use of the ajmaline test increases disease penetrance from 32.7% to 78.6%.
Molecular study results show that sodium channels with missense mutations inactivate faster in a temperature dependent way. In Dr. Brugada’s series of 723 patients, 364 have been screened for a mutation, with a mutation identified in 73 patients. The presence of a mutation does not always result in Brugada syndrome. Some mutation-positive family members had the classic Brugada ECG while others only had conduction defects.
In a long-term follow-up study of 361 of Dr. Brugada’s adult patients with Brugada syndrome, the mean age was 44.5 years, 285 (78.9%) were male, and 76 (21.1%) were female. Of the total, 58% were asymptomatic, 19% had syncope, and 22% had sudden death. The basal ECG was diagnostic in 288 patients (80%), while ECG with a Class I drug was diagnostic in 73 patients (20%). An implantable cardioverter defibrillator (ICD) was implanted in 188 patients (52%). During 65 ± 46 month follow-up, 116 patients (32%) had VF or sudden death. The men had a significantly higher incidence of these major events than the women (log-rank <0.003). The most events occurred in patients with previous aborted sudden death (log-rank <0.0001), followed by those with previous syncope, and the least occurred in asymptomatic patients. Patients who were inducible had more events during follow-up than those who were not inducible (log-rank <0.0001).
Multivariate analysis confirmed that the hazard ratio (HR) for sudden death was significantly higher for patients who were male (HR 2.13, p=0.01), those who had previous syncope (HR 3.77, p <0.001), those who had previous aborted sudden death (HR 5.28, p<0.001), and those who were inducible (HR 2.73, p=0.001).
Prevention of sudden cardiac death in pediatric patients with Brugada syndrome is a challenge due to the difficulty of implanting an ICD in a child and problems with follow-up. Dr. Brugada has followed 68 children with Brugada syndrome. The mean age at presentation was 11.8±4.7 years and 58.5% were males. About 75% had familial forms of Brugada syndrome, 66% were asymptomatic, 15% had syncope, and 19% had sudden cardiac death. The basal ECG was abnormal in 40% and normal in 60%, 13% were EPS inducible, 37% were noninducible, and 50% were not induced. An ICD was implanted in 17 children (25%).
During a mean follow-up of 44 months, 9 children (13.2%) had major events, including 3 sudden deaths and 6 ICD interventions. At any time during their lives, 18 patients had either sudden death or appropriate ICD therapies. Of these, 13 patients had aborted sudden death before diagnosis, with 5 of these having recurrent events treated by the ICD. Of 4 patients without previous sudden death, 2 experienced sudden death and 2 had appropriate ICD therapy. One patient presented with a VF episode during the ajmaline test. Analysis of these events showed a significantly higher risk of a major event in patients with previous aborted sudden death or syncope compared to asymptomatic patients (log-rank <0.001). Multivariate analysis showed that previous syncope (OR 15.9, p=0.017) and sudden death (OR 30.2, p=0.002) were independent risk factors for sudden death in this pediatric population.
Dr. Brugada concluded that Brugada syndrome in pediatric patients has a similar presentation as in adult patients, with the exception of the EPS value, which is probably due to the limited number of patients. Previous symptoms identify the majority of patients who will have events during follow-up, highlighting the need for aggressive therapy in these patients. Dr. Brugada closed his presentation by noting that each year there are 120 papers with Brugada syndrome in the title published and more than 1,500 citations dealing with Brugada syndrome. This shows how important Brugada syndrome has become to the scientific community.
|