A number of cytokines are increased in patients with heart failure. Although anti-cytokine therapy has been helpful in other diseases such as rheumatoid arthritis, it has proven to be a challenge in the treatment of heart failure. Dr. Kirk U. Knowlton, University of San Diego, discussed the effects of inflammation and the potential for anti-cytokine therapy in patients with heart failure.
According to Knowlton, understanding the etiologies of heart failure is key to understanding the disease process. From an immunologic perspective, certain cardiomyopathies are clearly initiated by inflammation, including viral infection and autoimmunity against the heart. Inflammation can also contribute to the progression of heart failure initiated by non-inflammatory causes, including ischemic coronary heart disease, valvular heart disease, and hypertension.
Studies have shown that myocarditis is worse in mice with severe combined immunodeficiency (SCID) that lack T cells, B cells, and immunoglobulins. With respect to innate immunity, coxsackievirus B3 (CVB3) myocarditis is worse when gp130 signaling is inhibited.
Although studies have shown that the immune response is beneficial in the early stages of viral infection, less is know about the effects of a chronic immune response. Levine et al (1990) demonstrated that TNF-a levels are markedly elevated in patients with severe chronic heart failure. TNF administration was shown to decrease cardiac function in rats; when the TNF was withdrawn, cardiac function improved. To explore this further, transgenic mice were generated that expressed cardiac specific TNA-a. These mice developed cardiomyopathy with increased end diastolic volume (EDV) and end systolic volume (ESV) and decreased ejection fraction (EF) compared to wild type mice. Additionally, myosin induced myocarditis has been demonstrated in the absence of viral infection, resulting in a marked cellular immune response directed against the myocardium.
Given the detrimental effects the immune system can have on the heart, it seems that immunosuppression would be beneficial in heart failure. In 1995, a trial was published in which patients with biopsy documented myocarditis were treated with immunosuppression. The results showed no significant differences in accumulated mortality between patients treated with immunosuppression and those who received placebo. There also was no significant difference in improvement of the EF over time with immunosuppression compared to placebo.
Since TNF-a antagonists have detrimental effects on the heart, several trials of TNF-a antagonist therapy for heart failure have been conducted. In the RECOVER and RENAISSANCE trials, patients with heart failure treated with etanercept had no significant improvement compared to those who received placebo. The RENEWAL trial had similar results, with no statistically significant difference in event-free survival between the etanercept group and the placebo group. The ATTACH trial studied different doses of infliximab compared with placebo. Patients who received 10 mg/kg of infliximab had a markedly increased rate of death and hospitalization due to worsening heart failure than those who received 5 mg/kg of infliximab or placebo. There was no difference in adverse events between the low-dose infliximab and placebo groups.
Given the failure of these trials, Knowlton suggested consideration of other innate immune signaling mechanisms that might induce or worsen cardiomyopathy, including transforming growth factor beta (TGF-b), interleukins, and TLR agonists. It also may be the case that too much or too little cytokine signaling may be detrimental, depending on the etiology of heart failure. Studies conducted in Knowlton’s laboratory have demonstrated the importance of appropriate regulation of cytokine signaling. For example, in viral infections, appropriately regulated gp130 signaling in the myocyte can be helpful while excessive gp130 signaling can be harmful.
|
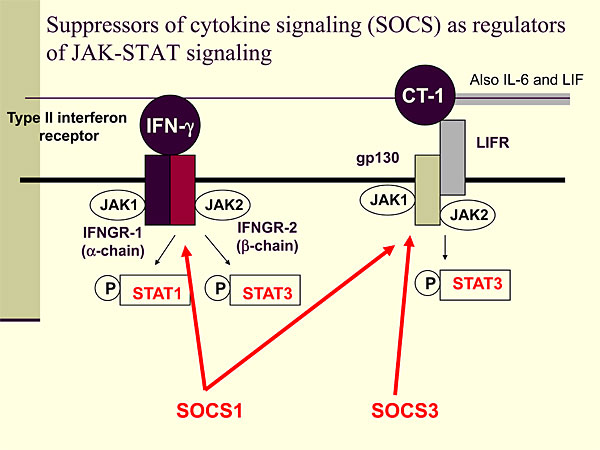
Figure 1. Suppressors of cytokine signaling (SOCS) as regulators of JAK-STAT signaling.
【Click to enlarge】 |
|
|
To further explore cytokine regulation in heart failure, Knowlton conducted studies of SOCS as regulators of JAK-STAT signaling (Figure 1). When an interferon agonist binds to a type II interferon receptor, JAK1 and JAK2 are activated, leading to phosphorylation of STAT1 and STAT3. When cardiotropin-1 (CT-1), IL-6, or leukemia inhibitory factor (LIF) binds to the LIF receptor (LIFR), a cascade is activated that leads to STAT3 phosphorylation. IFN-g activates SOCS1, which can inhibit interferons and CT-1, along with gp130 signaling. SOCS3 is induced by gp130 signaling and can inhibit gp130 with some specificity but has no effect on interferon signaling.
|
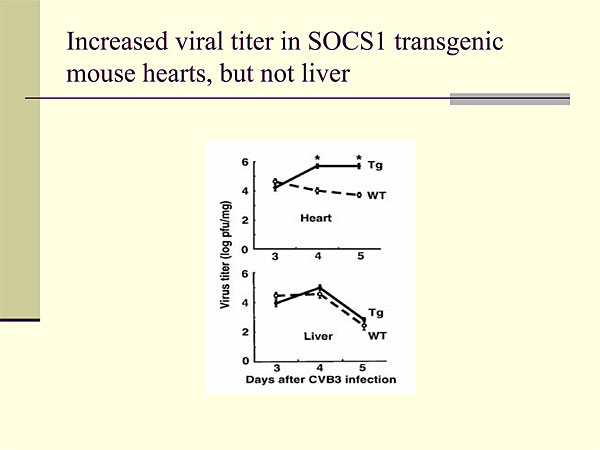
Figure 2. Increased viral titer in SOCS1 transgenic mouse hearts, but not liver.
【Click to enlarge】 |
|
|
To study this further, Knowlton generated transgenic mice that expressed SOCS1 and SOCS3 molecules with the a-myosin heavy chain promoter. The mice had normal hearts and a normal response to pressure overload before infection. They were infected with CVB3 and analyzed for four to five days post infection. Infection with coxsackievirus is known to induce JAK-STAT signaling in the heart. Analysis of the SOCS1 mice showed that cardiac specific expression of SOCS1 markedly increased CVB3-mediated cardiac injury compared to wild-type mice. This was associated with an almost 100-fold increased viral titer in the SOCS1 mouse hearts but not in the livers (Figure 2).
Previous experiments with interferon receptor deficient mice showed that global knockout of interferon receptors has no effect on viral infectivity in the heart, indicating that endogenous interferon does not have an important role in antiviral mechanisms in the heart.
SOCS3 does not inhibit interferon signaling but has a major effect on gp130 signaling. Like the SOCS1 transgenic mice, the SOCS3 mice had markedly increased cardiac injury after CVB3 infection compared to wild-type mice. At day 4 after infection, there was no evidence of a cellular inflammatory response in the SOCS3 mice. Quantitative analysis showed a 100-fold increase in virus titer in the heart but not in the liver. There also was an increase in early mortality, even prior to onset of a cellular immune response (p <0.0001). The SOCS3 mice had increased left ventricular end systolic dimension and decreased fractional shortening compared with wild-type mice.
These experiments showed that forced expression of SOCS1 or SOCS3 have no effect on normal cardiac growth and function. Overexpression of SOCS1 and SOCS3 markedly increases the susceptibility to CVB3 infection in the intact heart. Expression of SOCS3 increased susceptibility to CVB3 in an interferon-independent manner.
To determine the role of gp130 signaling, cardiac-specific gp130 knockout mice were infected with CVB3 and analyzed 120 hours post-infection. The knockout mice had an increased susceptibility to viral infection compared to wild-type mice, although to a lesser extent than seen in the SOCS1 and SOCS3 transgenic mice. This suggests that gp130 is important but may not be the only mechanism by which SOCS3 increases susceptibility in cardiac myocytes. Further studies suggested that one of the mechanisms by which gp130 signaling inhibits susceptibility to viral infection is by stabilizing and increasing the expression of dystrophin glycoprotein complexes within the sarcolemma.
To determine the effect of SOCS3 knockout in the heart, cardiac-specific SOCS3 knockout mice were generated that have high efficiency SOCS3 knockout in the cardiac myocytes. Preliminary findings of this ongoing study show that the SOCS3 knockout mice developed dilated cardiomyopathy with increased left atrial size, left ventricular wall thinning, and increased chamber size compared to wild-type mice. The SOCS3 knockout hearts also had a marked decrease in fractional shortening and high mortality compared to the controls. The SOCS3 knockout hearts had little or no increase in fibrosis and normal sarcomeric organization. STAT3 and ERK1/2 phosphorylation were present at 15 weeks of age. There also was an increase in gp130, Akt, and Jnk downstream signaling. Additionally, some data show that there was at least partial rescue with heterozygous gp130 cardiac-specific knockout mice, demonstrating a gp130 signaling mechanism is the likely cause.
Knowlton concluded that the immune response likely has both good and bad influences on the myocardium. However, researchers have a long way to go before learning how to effectively modulate immune function to benefit patients with heart failure. Inhibition of the renin-angiotensin system (RAS) has positive effects on essentially all forms of heart failure, but such may not be the case for inhibition of an immune process that is important for controlling infectious causes of heart failure. |