The importance of employing molecular genetic testing to distinguish phenotypes within cardiomyopathies, based on the understanding of the molecular genetic basis of hypertrophic, dilated, and right ventricular cardiomyopathies from research over the last two decades, was stressed in this lecture by Professor William J. McKenna. Within hypertrophic cardiomyopathy (HCM) where differentiation can be made this is particularly important. Gene mutations that encode sarcomeric contractile proteins (SCP) have been identified and are likely the major cause of hypertrophic cardiomyopathy (HCM). Yet, there are persons who are diagnosed with HCM without SCP disease-causing mutations. In conditions like right ventricular cardiomyopathy, improved mutation analysis to better understand the phenotype and prognosis is needed.
Incomplete disease expression in HCM is common
The phenotype of HCM is characterized by asymmetric hypertrophy, left ventricular outflow tract (LVOT) obstruction, and the typical abnormal electrocardiogram. Histologic features include monocyte disorganization surrounding increased areas of loose connective tissue.
Importantly, incomplete disease expression is seen in a significant proportion of patients, if not a majority, who carry SCP gene abnormalities, including HCM, right ventricular myopathies, and ion channel diseases. These patients do not exhibit the typical asymmetrical hypertrophy or LVOT, and their histologic features are unknown.
|
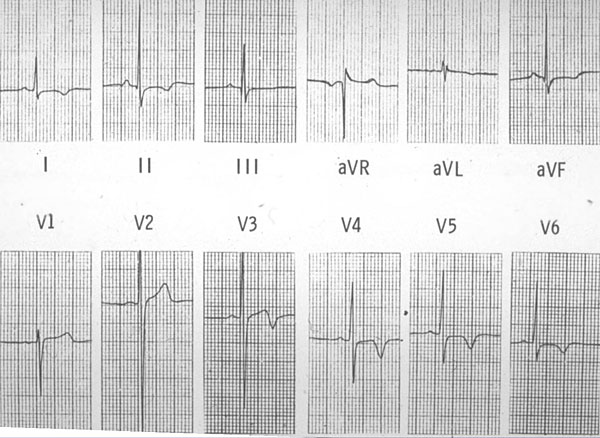
Figure 1. ECG in a woman with cardiac troponin T disease shows left ventricular hypertrophy (LVH) with repolarization changes, although clinical LVH was not present.
【Click to enlarge】 |
|
|
A striking example of this incomplete expression is a family presented by McKenna with cardiac troponin T disease with myocardial disarray and premature sudden death in four family members (aged 21, 23, 38, and 42 years), and it highlights the need for molecular genetic analysis. Notably, heart weight and wall thickness were normal in those who died, thus the typical echo features used for clinical diagnosis may not be seen, despite the presence of the typical histologic features. The Echo of the presenting asymptomatic 23 year-old woman was completely normal, but her 12-lead ECG showed left ventricular hypertrophy (LVH) with repolarization changes, although she did not have clinical LVH (Figure 1).
Unexplained left ventricular hypertrophy
Genetically-determined forms of metabolic storage diseases or other genetically-determined syndromes are found in a significant proportion of patients with so-called unexplained LVH who historically were labeled as having HCM. Correct diagnosis is important, because of differences in prognosis, management, genetics, and genetic counseling nowadays.
|
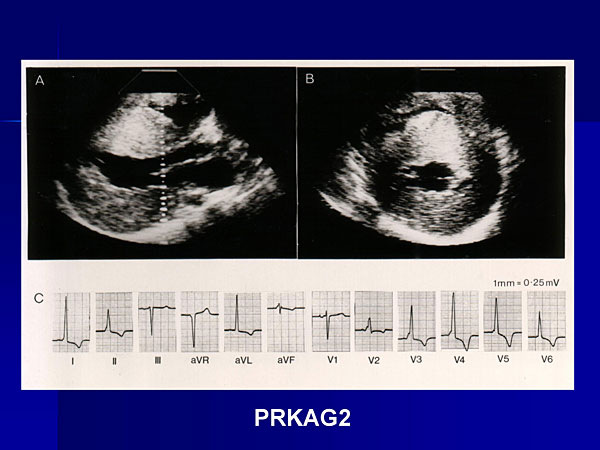
Figure 2. Evidence of pre-excitation was seen in a young man later identified to have a mutation in AMP kinase.
【Click to enlarge】 |
|
|
McKenna presented a representative case, a young man first seen in 1978 at the age of 12 years, when he had 3-4 cm of LVH and about 1 cm of right ventricular hypertrophy (RVH), and evidence of pre-excitation (Figure 2). He died suddenly from an exacerbation of heart failure. Analysis of stored blood samples for genetic screening identified a mutation in AMP kinase, a phenotype with premature conduction disease, accessory pathways, and perhaps severe hypertrophy and in some patients skeletal muscle abnormalities. McKenna noted that a correct diagnosis probably would not have influenced medical management in this patient.
|
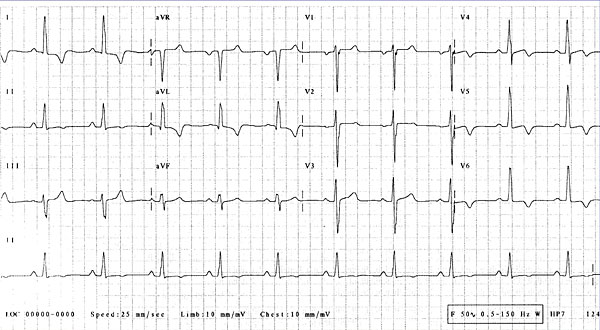
Figure 3. An ECG in a 60 year-old woman later identified as having Fabry’s disease shows LVH with repolarization changes.
【Click to enlarge】 |
|
|
|
Figure 4. An Echo in a 60 year-old woman later identified as having Fabry’s disease revealed asymmetrical septal hypertrophy with about 1.6 mm wall thickness.
【Click to enlarge】 |
|
|
|
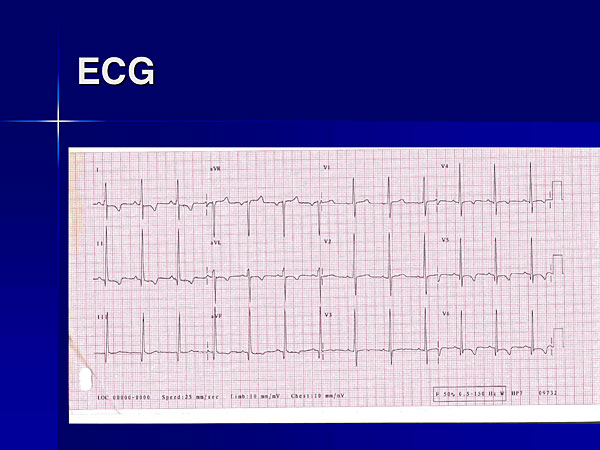
Figure 5. A patient with a mitochondrial abnormality had LVH with repolarization on ECG.
【Click to enlarge】 |
|
|
| |
In contrast, another example in which Fabry’s disease was ultimately diagnosed highlights how a correct diagnosis may have saved lives. The presenting patient was a 60-year-old woman whose abnormal 12-lead ECG was attributed to coronary artery disease. Significant family history included a sister diagnosed with HCM at the age of 59, two brothers who died from cardiac disease in their 60s, and early onset deafness in her mother, sister, and daughter; and a nephew with end-stage renal failure and hypertension at a young age. LVH with repolarization changes was seen on ECG, but nothing specific in relation to conduction, accessory pathways, or pattern of repolarization disease (Figure 3). Her Echo revealed asymmetrical septal hypertrophy with about 1.6 mm wall thickness (Figure 4). DNA analysis confirmed a disease-causing mutation (N215S Heterozygote). Enzyme replacement therapy, although likely to have little influence on HCM, may have influenced other aspects of Fabry’s disease that manifested in this family.
A mitochondrial abnormality was identified in another family presented by McKenna, again highlighting the value of correct diagnosis for management and genetic counselling. A 41-year-old woman referred for cardiac transplantation had been symptomatic from age 13, including exercise intolerance, exertional breathlessness, and muscle aches, and migraines. Her monozygotic twin had identical symptoms, mild LVOT obstruction, a pacemaker, and had been considered for myectomy. The patient had LVH with repolarization on ECG (Figure 5) and mild asymmetrical septal hypertrophy on Echo. On metabolic exercise testing, her peak oxygen consumption was 10.5 ml/kg/min, only 27% of predicted for her age and size, and metabolic acidosis apparent within 30 seconds of starting exercise. Treatment included high-dose riboflavin, which increased her peak oxygen consumption to 15 ml/kg/min.
ARVC: Right ventricular cardiomyopathies
Features of arrhythmogenic right ventricular cardiomyopathy (ARVC) include cell adhesion, myocytes coming apart, and fatty and fibrous cell replacement. The disease appears to develop from the epicardium to the endocardium, with relative sparing of the endocardium. Early clinical diagnosis is difficult, with no single test being definitive, and Echo and MRI being limited because they only show late manifestations. Molecular genetic diagnosis is particularly important in ARVC
|
Figure 6. The natural history of arrhythmogenic right ventricular cardiomyopathy.
【Click to enlarge】 |
|
|
The natural history of ARVC is shown in Figure 6. Sudden death occurs during the concealed phase, and the end stage, where severe left or right ventricular impairment can occur with complications, such as atrial fibrillation, emboli and heart failure, is not well recognized. The overt arrhythmic phase (occurring in the teens, young adult years) is the recognized phase, characterized by symptomatic ventricular arrhythmias. Notably, arrhythmic risk can occur when mechanical and functional changes in the RV are not apparent.
The identification of five cell adhesion disease-causing genes, all related to desmosomal function and mechanical integrity of the cell junction, led to the understanding of ARVC. Defects in these genes (desmoplakin, desmoglein, desmocollin, plakophilin, and plakoglobin) cause electrical abnormalities leading to ECG changes, arrhythmias, and sudden death at a stage where the mechanical defect has very mild consequences and is not seen on Echo or MI.
Genetics was important to recognize LV arrhythmogenic cardiomypathies, where the predominant manifestation of the cell-adhesion disease-causing gene is in the LV, with a LV pattern on ECG showing T wave conversion in V3 to V6. RV volumes and overall function of RV and LV are completely normal on MRI, and on Echo the LV volumes and systolic thickening are normal. However, a very abnormal pattern of mid-myocardial late enhancement consistent with the ECG was seen on late enhancement, revealing the origin of the arrhythmia. A desmoplakin mutation was the cause in 9 of the 12 affected family members with predominant LV involvement. McKenna noted that although how common this is, examination of families with more typical ARVC has revealed this more predominant LV manifestation in some persons.
|