Cardiac remodeling is influenced by six molecular pathways in addition to hemodynamic load and neurohormonal activation. These pathways provide potential therapeutic targets for heart failure (HF). In this presentation, Dr. Eun-Seok Jeon, Samsung Medical Center, Korea, reviewed studies of these molecular pathways and targets.
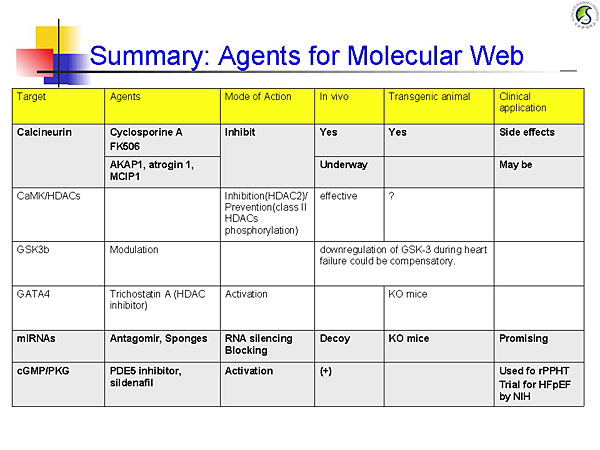
Calcineurin dephosphorylates NFAT, which translocates to the cell nucleus and activates genes that cause hypertrophy. Endogenous calcineurin inhibitors–AKAP1, atrogin 1 (FBXO32), and RCAN1– are being studied as inhibitors of this pathway. Calmodulin and its kinase contribute to cardiac hypertrophy through HDAC and RyR phosphorylation.
Glycogen synthase kinase 3b (GSK3b) normally inhibits hypertrophy but is suppressed by Akt or PKA phosphorylation, allowing progression of hypertrophic pathways. Experiments in mouse models showed that persistent GSK3b inhibition induces compensatory hypertrophy, inhibits apoptosis and fibrosis, and increases cardiac contractility.
Deletion of the transcription factor GATA4 in mouse models results in progressive cardiac dysfunction and dilation, with pressure overload causing rapid decompensatory HF. GATA4 regulates cardiac gene expression, hypertrophy, stress compensation, and myocyte viability.
MicroRNAs (miRNAs) mediate post-transcriptional gene silencing. In vitro overexpression of miRNA-133 inhibited cardiac hypertrophy. In vivo miRNA-133 inhibition caused marked, sustained hypertrophy. MicroRNA-208-/- mice had reduced hypertrophy in response to pressure overload. These data show that these miRNAs are key regulators of cardiac hypertrophy.
Cyclic GMP (cGMP)-dependent protein kinases regulate phosphodiesterases (PDE). The PDE5A inhibitor sildenafil suppresses hypertrophy and improves heart function in mice with induced pressure overload and reverses pressure overload-induced hypertrophy. An ongoing NIH-sponsored trial is testing PDE5 inhibitor therapy for HF with preserved EF.
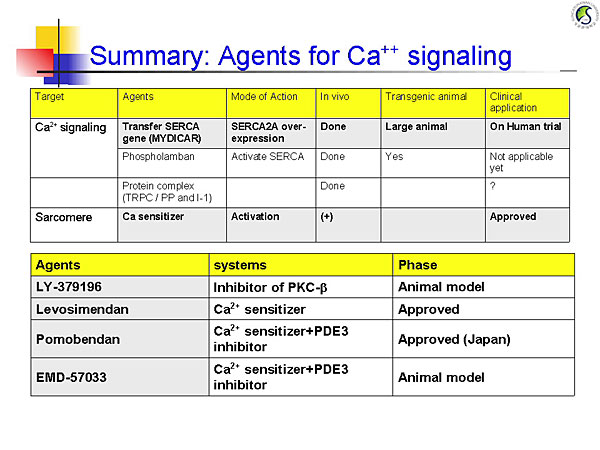
Ca2+ is the central regulator of excitation-contraction coupling. Potential therapies in calcium signaling pathways include SERCA2A gene transfer, phospholambam gene deletion, and production of constitutively phosphorylated phospholambam, all of which improved heart function and remodeling in animal models. A phase 1/2 trial is investigating AAV1/SERCA2a gene transfer in patients with NYHA III/IV HF with EF ≤30%. The initial outcomes of this trial will provide valuable data for molecular targeted therapy for HF.
|
Figure 1. Agents for the molecular web
【Click to enlarge】 |
|
|
|
Figure 2. Agents for calcium signaling.
【Click to enlarge】 |
|
|
Calcium sensitizers stimulate cardiac contractility without causing intracellular calcium overload or increasing myocardial oxygen demand. Mutations in the titin cap (T-cap) gene that promote association of T-cap with titin and calsarcin 1 cause hypertrophic cardiomyopathy. Mutations that impair this association cause dilated cardiomyopathy. Manipulating these interactions might provide a new therapeutic approach for HF.
Investigation of novel agents targeting molecular pathways (Figure 1) and calcium signaling pathways (Figure 2) is promising for the treatment of HF.
|