In 1972 Eugene Braunwald proposed that interventions should be found to protect the ischemic heart from infarction. His idea was to develop a drug that when used with reperfusion, would reduce the amount of infarction and complications. Yet, 35 years later the only treatment available for acute myocardial infarction (AMI) is reperfusion therapy. In this lecture, Dr. James Downey, University of South Alabama, discussed the history and issues of cardioprotective drug research.
During the 1980s and 1990s drugs were tested based on popular theories of causes of ischemic myocardial death. Subjects of investigations included free radical scavengers, beta blockers, calcium antagonists, and anti-inflammatory agents. All of these were controversial in preclinical studies and ineffective in clinical trials. For example, one laboratory conducted a series of animal studies with low-dose adenosine at reperfusion, all of which had positive results. However, other laboratories were unable to duplicate these results.
These preclinical studies of low-dose adenosine were followed by two AMISTAD clinical trials, which also had equivocal results despite large numbers of patients. AMISTAD1 was only able to show positive results in a retrospective subgroup analysis of anterior myocardial infarction. Applying the same analysis to the larger AMISTADII failed to demonstrate significant myocardial protection. Dr. Downey recommended that before such clinical trials are begun, any discrepancies among the preclinical trials should be resolved.
A 1986 study reported that dogs subjected to 40 minutes of coronary occlusion followed by reperfusion had 25% infarction of the ischemic zone. Dogs preconditioned with four 5-minute periods of occlusion followed by reperfusion had only about 10% infarction. All subsequent animal studies were able to reproduce these results. However, ischemic preconditioning could not be translated to the clinical setting.
|
Figure 1. Preconditioning Mechanism.
【Click to enlarge】 |
|
|
The mechanism of preconditioning starts with the trigger pathway that occurs during the short periods of ischemia and reperfusion before the lethal ischemic insult (Figure 1). Signal transduction pathways beginning at the opioid, bradykinin, and adenosine receptors cause opening of the potassium channel in the mitochondria, producing free radicals that activate protein kinase C (PKC). The mediator pathway occurs at reperfusion after the lethal ischemic insult and involves ERK, Akt, and other signal transduction elements. This pathway ultimately inhibits the mitochondrial permeability transition pore (mPTP), killing the mitochondria in the first few minutes of reperfusion. This inhibition of the transition pores is what provides protection. Because protection occurs at reperfusion, ischemic and pharmacologic postconditioning can be given at reperfusion to provide protection through the mediator pathway.
In 2003, a study by Zhao et al showed that in dogs inhibition of infarction by ischemic postconditioning during reperfusion provided protection as potent as ischemic preconditioning. Pharmacologic agents that can provide postconditioning protection through the mediator pathway include natriuretic peptides, guanylyl cyclase activators, erythropoietin, growth factors, cyclosporine, subtype-selective adenosine agonists, intracoronary bradykinin, and delta opioid agonists. These agents have been proven effective in all the animal species tested and are currently entering clinical trials.
Another issue is whether cardioprotectants are needed. Some experts believe that a therapeutic ceiling has been reached with current reperfusion therapy and that no additional benefit will be gained with additional cardioprotection. A 2008 review (Miura et al) of 10 studies found that about 50% of ischemic tissue survives with reperfusion therapy alone. Evidence shows that symptoms appear when the overall infarct size involves more that 20% of the left ventricle. Among the entire population that receives reperfusion therapy, infarct size is kept below 20% in about 75% of patients. Thus, these patients would not benefit from a cardioprotective drug, but the remaining 25% would.
Clinical trial design is an important consideration in cardioprotectant investigations. A major problem is that the 75% of patients who would have had small infarcts without the drug dilute the data. This issue is especially important when clinical outcomes are used as the endpoint. An example is the J-WIND trial of atrial natriuretic peptide (ANP) published in 2007. In this study, the high-risk 25% of patients were not identified. Infarct size was reduced by 14.7% and ejection fraction improved by 1.7 points in patients treated with ANP (N=569) versus controls. These results were statistically significant but clinically insignificant.
|
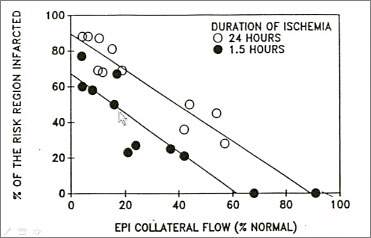
Figure 2. Factors Determining Infarct Size in Laboratory Animals
【Click to enlarge】 |
|
|
Three factors determine infarct size in laboratory animals—size of the ischemic zone, level of collateral flow, and duration of ischemia (Figure 2). Michel Ovize found that infarct size in control patients was not correlated with ischemic time (duration of symptoms) or collateral flow (angiographic evidence) but was correlated with risk zone size. Piot et al found that when risk zone size is included in the analysis, the 25% of patients who need a cardioprotectant can easily be identified. This does not mean that treatment should only be given to those with large risk zones. According to Dr. Downey, if the treatment is effective for the high risk group, innocuous, and inexpensive, then all should be treated to benefit the few who actually need it.
|
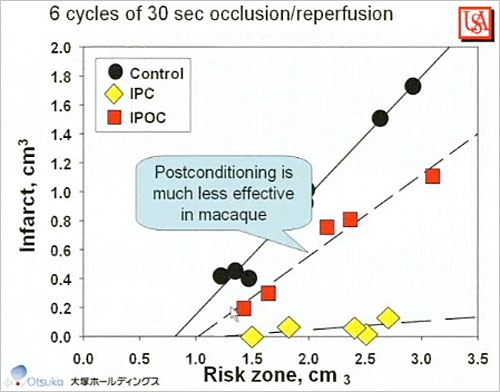
Figure 3. Postconditioning in Macaque Monkeys
【Click to enlarge】 |
|
|
Animal hearts may not be an appropriate model of the human heart. Whereas in rabbits reperfusion may be given after 30 minutes of ischemia, time to reperfusion in human patients is 180 minutes or longer. Gersh et al concluded that the ultimate human infarct size is reached 5 hours after occlusion versus 1 hour in rabbits. In macaque monkey studies, Dr. Downey found that: 60 minutes of ischemia did not produce infarction but 90 minutes produced 40% infarction; collateral flow (6% of normal) is too low to significantly protect the macaque heart; ischemic preconditioning significantly protects macaque hearts from infarction (p<0.001); and infarct size is predictable. However, postconditioning was much less effective in macaques than in other animal models (Figure 3).
Animal studies typically have been performed in healthy animals while most AMI patients have many co-morbidities. Studies in aged animals and those with diabetes indicate that co-morbidities may decrease the effectiveness of ischemic preconditioning. Further investigation is needed into which co-morbidities interfere with cardioprotection and how that can be overcome.
Dr. Downey concluded that: The wrong drugs have been tested as cardioprotectants; a therapeutic ceiling has not been reached with current reperfusion therapy; clinical trials have been inadequately designed; animal hearts probably are not an appropriate model of the human heart; and co-morbidities may blunt the actions of cardioprotectants.
|