Genomics is on the cusp of changing all
of biology, making this a very revolutionary time. An
overview of genomics, methodology for expression profiling,
and examples of its application in cardiovascular medicine
were provided in this lecture.
|
PAGE
TOP
|
Function genomics is the study of
individual genes, proteins, or pathways within the
broad context of the genomics of the cell, tissue
or organisms. Specific functions are considered
within a large percentage or even the entirety of
the genome of that cell.
Functional genomics is important for
the new biological insights it provides and because
it may yield the next major discoveries in pharmaceutical
therapeutics. Genomics will shorten the painstaking
10-year process usually required for lead optimization
to about 3-4 years. Using the genome will shorten
the process of target identification (characterizing
genes as candidates, using disease tissue, cellular
models, and animal models), target validation (discrimination
of valid targets relevant to the disease), lead
identification (using compound screening with high
throughput screening) and lead optimization (search
for optimal efficacy in further studies or clinical
studies).
|
|
PAGE
TOP
|
The Human Genome Project encompasses
a large number of goals, particularly the genetic
and physical map that is nearing completion. A great
effort has also been made in the areas of technology
transfer and informatics, which will be the keys
over the next decade to exploiting all of this information.
Efforts have also been made in the areas of education
and ethics. The Human Genome Project began in 1990
and is now an international collaborative year.
The project goals are to:
- Identify all (> 100,000 genes) in human DNA
- Determine the 3 billion chemical base pairs
of human DNA
- Store this information in public databases
- Develop tools for data management and analysis
- Address the ethical, legal and social issues
The Human Genome Project reported
in Nature in December 1999 the sequencing
of the 33.5 million base pairs of chromosome 22.
They reported there were more than 545 genes, more
than 134 pseudogenes, 39% of the DNA was copied
into RNA, only 3% was made into protein, 247 known
genes, 150 genes had some homology to genes in the
database, and 148 expressed sequence tags. As of
April 2000, 65% of the genome is sequenced and a
90% rough draft will be available in June 2000,
with completion by the end of 2000.
|
|
PAGE
TOP
|
A large number of expressed sequence
tags (EST) or cDNA tags are now stored in databases.
In the public database there are about 70,000 EST
and the proprietary Incyte database claims to have
about 90% of the genome completed. It is likely
that about 10% of EST are missed in the gene libraries
because some genes are probably expressed in very
low abundance and some are temporally expressed
only in specific tissue. This is likely to be an
important factor as it is quite possible that therapeutic
targets will be rare genes or temporally expressed
genes, because therapies directed at such genes
may have the least side effects. Therefore, while
there is a bias towards knowing more in the early
period, there is also a bias towards possible golden
material coming at the end of the EST database.
The number of gene patents in the
US alone is skyrocketing, due to efforts to protect
various amounts of data. The uses of EST have ranged
from efforts to patent the individual sequence alone,
to more recent efforts to obtain a patent for the
function and possible use of the EST information.
Interestingly, while the patents are increasing,
the function of only about 10-15% of the current
EST database is known. Rough homologies can be drawn
for about 50% of the genes, but at least 50% of
the genes have no known function.
|
|
PAGE
TOP
Gene tissue
expression patterns |
|
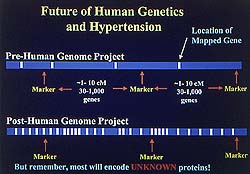 |
Figure
1. Most newly identified genes will encode for
proteins with no known function.Expression
patterning will work with genetic mapping to allow
faster identification of gene function. (Lee 2000)
Click to
enlarge |
|
The dense genetic map and the advent
of single nucleotide polymorphisms available throughout
the genome will speed the identification of the
gene of interest. This will simplify the genetic
analysis of complex, polygenic diseases, which was
previously accomplished by positional cloning.
Although epidemiologists and molecular
epidemiologists are excited about this new information,
it must be remembered that even with this new ability
most of the genes will encode for proteins for which
there is no known function, as illustrated in Figure
1. If this is true, it will be important that much
of the expression pattern data is available to the
public to narrow the analysis to the specific gene
that may be causative in a disease by looking at
the information related to the gene expressed in
the disease or tissue of interest. Expression patterning
will work together with genetic mapping to allow
faster identification of gene function.
Tissue expression methodology
Past methods for defining expression
profiles have been slow and tedious, including the
use of subtractive hybridization, differential display,
and mass sequencing and serial analysis of gene
expression techniques. This will be replaced by
hybridization arrays, which are the most efficient
and will be the least expensive ways to perform
expression profiling in the laboratory.
|
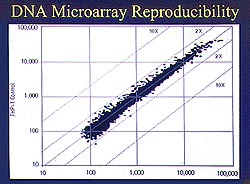 |
Figure
2. A DNA microarray reproducibility experiment
shows that the microarray is equivalent to performing
a Northern Blot on 10,000 genes simultaneously.
(Lee 2000)
Click to
enlarge |
|
A genetic microarray is the analysis
of up to 20,000 genes simultaneously. In the Synteni
technique one form of RNA is labeled with a red fluorescent
marker and the other with green which are then hybridized
and read by a laser scan for the differential expression.
There are internal controls, but there is a single
hybridization for each single gene of interest. A
DNA microarray reproducibility experiment shows that
the microarray is equivalent to performing a Northern
Blot on 10,000 genes simultaneously (Fig. 2). The
reproducibility has been quite good and the quantitation
has been remarkable, particularly compared with techniques
such as differential display.
The Affymetrix system is a fascinating
system in which photolithography is used to synthesize
the oligonucleotide sequences on the chip in solid
phase. It is very sensitive and specific quantitatively,
which is important as about 20 hybridizations are
done for each gene of interest and 20 controls.
The advantages of this system is that it is truly
"plug and play", extremely specific, highly sensitive,
and interfaces with bioinformatics. It is high density,
due to the photolithography technique, and can analyze
about 100,000 genes per single chip. By the time
the genome sequencing is completed, there is the
potential for analyzing every gene on one chip.
The disadvantages are that making the photolithography
mask is expensive and tedious, making the chip itself
very expensive. The cost per chip has fallen from
about $10,000 to $1000-2000. The Affymetrix chip
can not be customized for a single laboratory.
In-house development of chips is now
possible due to an evolution and explosion of the
technology. Individual laboratories may find this
daunting, but individual research centers can undertake
this. About 20 different companies provide the technologies
to make this possible. The advantages are lower
production costs (after the cDNA purchase) and custom
design to a particular diagnostic problem. Disadvantages
are the initial set-up, the initial cost of the
cDNA, errors in EST database or cDNA suppliers that
may require mass sequencing and validation steps,
and a loss of sensitivity and specificity compared
to the Affymetrix technique.
|
|
PAGE
TOP
Application
of tissue expression |
|
|
|
Tissue expression will have remarkable
utility. Tissues or cells may be input to look at
the effects of compounds or inhibitors or specific
mutations that may participate in pathways, resulting
in outputs that may include prognosis for cancer,
diagnosis of different types of tumors, therapeutic
targets for drug validation, efficacy, and toxicity.
Pharmaceutical companies are beginning
to use this ability to determine toxicity by mapping
out the hepatic pathways for different types of
toxins. With this technology it is possible to visualize
that particular compounds are toxic in rat livers
at a very early stage, for example, well before
extensive toxicology studies with hundreds of animals.
In these days of mass high throughput chemical screening
this analysis is very useful before spending lots
of money.
Application in cardiovascular medicine
Lee used transcriptional profiling,
or transcript imaging, to map a picture of an area
that is not well understood: the molecular response
of cells in human atherosclerotic lesion. The results
obtained were highly specific, all of which were
confirmed by multiple Western and Northern analyses.
Clusters of functions seemed to be induced by the
particular stimuli. The small number of genes induced
simplified potential identification of physiologically
relevant genes. This was a special circumstance,
as usually in the lab a larger portion of the genome
is being affected.
|
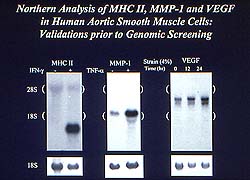 |
Figure
3. Free stimuli of interferon, tumor necrosis
factor (TNF) and mechanical strain were studied
using Northern blot analysis as a control. MHC
II was robustly induced by interferon, TNF induced
MMP-1, and vascular endothelial growth factor
(VEGF) was mechanically induced. (Lee 2000)
Click to
enlarge |
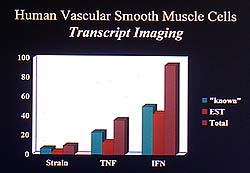 |
Figure
4. Mechanical deformation interestingly led to
a very small number of induced genes, compared
to the interferon and growth factor stimuli. (EST,
expressed sequence tag; TNF, tumor necrosis factor;
IFN, interferon.) (Lee 2000)
Click to
enlarge |
|
In this work, Lee looked at different
pathways that might affect atherosclerotic lesion
stability in human aortic smooth muscle cells using
genomic screening. Free stimuli of interferon, tumor
necrosis factor (TNF) and mechanical strain were studied
using Northern blot analysis as a control (Fig. 3).
MHC II was robustly induced by interferon, TNF induced
MMP-1, and vascular endothelial growth factor (VEGF)
was mechanically induced. Good internal controls are
needed with these genetic studies to save time and
money and confirm good specimens. In the 10,000 gene
array, dozens of genes were induced by TNF, including
a number that were already known such as MCP-1, superoxide
dysmutase, IL-4, VCAM, and ICAM-1.
Interferon gamma was even more prolific
and induced a large number of genes, many known
to be involved in vascular biology in response to
interferon and a number involved with interferon-induced
apoptosis in other cells (MHC Class II, ICAM-1,
MCP-1, SOD, and ICE). Interestingly, mechanical
deformation led to a very small number of induced
genes, compared to the interferon and growth factor
stimuli (Fig. 4). The types of genes induced were
quite specific. There was downregulation of MMP-1
(leading to collagen accumulation) and upregulation
of PAI-1 (leading to decreased matrix degradation);
both genes participate in intracellular matrix degradation.
Also present was upregulation of VEGF and upregulation
of cyclooxygenase-1 leading to increased prostacyclin.
Other use of transcriptional profiling
include quality controls for primary cell populations
to rule out false positives and determine reproducibility.
One of the advances of bioinformatics is performing
mathematical clustering techniques using the same
types of analyses used for multivariate epidemiology.
Those techniques are moving closer to being able
to identify common pathways between cells. In this
fashion a molecular map can be developed in which
all information is integrated through the genome
into common pathways that can be understood. This
illustrates the need for information to be shared
and why information technology is needed to share
information from the different types of experiments.
|
|
PAGE
TOP
|
Expression patterns will be pursued,
and meanwhile classical biochemistry and cell biology
will continue as it is the basis. In vivo validation
using transgenics and knock-outs is a critical factor.
Exploiting gene transfer and integrative physiology
further with techniques such as high throughput
yeast to hybrid screenings is needed.
A summary of the known protein or
gene targets for all of the therapies currently
in use shows there is about 483 targets. About 45%
of the targets are receptors and 28% are enzymes.
The pharmaceutical industry estimates using high
throughput screening there are 4000-4500 different
small molecule genes that are targetable. Over the
next few years the data to increase these targets
10-fold will become available. This ability is unprecedented
and an exciting part of the entire genome project.
Microarray research must be just as
robust and thoughtful as classical hypothesis-driven
research. It can not rescue a bad model, bad experiment
or design. With microarray research an hypothesis
is researched across a large portion of or the entire
genome. It is important to consider that taking
great care in these experiments can save years of
chasing false leads, Good controls, a great deal
of care, reproducibility, the same sort of thought
that goes into all experiments, are the key items
in microarray experiments as well.
|
|
PAGE
TOP
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2000
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|