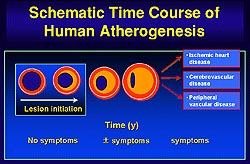
The presence of the mature atherosclerotic
lesion sets the stage for complications of atherosclerosis.
Patients may present with symptoms from stenotic
lesions or with thrombotic symptoms, such as acute
myocardial infarction or unstable angina, which
may occur without warning as in chronic stable syndromes
of atherosclerosis.
Much has been learned about the pathogenesis
of the thrombotic complications of atherosclerosis.
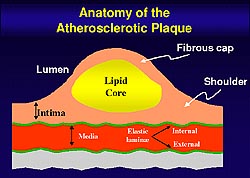
The fibrous cap of the plaque protects the integrity
of the atheroma, providing a shield between the
thromogenic material in the plaque's lipid core
and the coagulation factors present in the circulating
blood. Weakening of the fibrous cap leads to plaque
rupture. The thrombogenicity of the plaque's lipid
core also plays a key role in the unstable coronary
syndrome.
Libby reviewed some of the thinking
and experimental results advocated by his laboratory
that helps to explain the features of the unstable
atheroma. When inflammation is present in the intima
the leukocyte can send signals to the smooth muscle
cell to inhibit the biosynthesis of collagen, which
is the key structural component of the plaque's
fibrous cap. The leukocytes can also exchange signals
that cause overexpression of collagen-degrading
proteinases. Thus, inflammation places the collagen
in the plaque's fibrous cap under the double attack
of decreased synthesis and increased breakdown,
setting the stage for plaque rupture and thrombosis.
The leukocytes can also exchange signals that can
augment the production of the procoagulants that
make it dangerous for the blood to enter the artery
wall.
In an experiment with human smooth
muscle cells in culture to measure collagen biosynthesis
it was shown that the smooth muscle cells incorporate
proline, an amino acid rich in collagen, in the
basal state into newly synthesized collagen. When
the smooth muscle cells are exposed to platelet-derived
growth factor (PDGF) or transforming growth factor-beta,
which are released during coagulation, there was
an increase in the biosynthetic rate of collagen.
For lesion healing this is very important. But,
when smooth muscle cells are exposed to gamma interferon,
a T-cell derived cytokine, new collagen synthesis
by smooth muscle cells was nearly inhibited. It
is now recognized that there are many T-cells in
various regions of the plaque, particularly those
prone to rupture.
Role of gamma interferon
Van der Wal observed some years ago
that in human fatal thrombotic events in coronary
arteries, macrophages and T-lymphocytes are the
dominant cell types whether or not there is thrombosis
due to plaque rupture or superficial erosion. Further,
the cells in that region overexpressed HLA-DR, a
transplantation antigen. Libby's laboratory defined
that antigen more than a decade ago as a gamma interferon-inducible
structure on the surface of the smooth muscle cell.
So, the recent clinical finding that there are HLA-DR-positive
smooth muscle cells at the sites where plaque ruptures
is strong evidence of gamma interferon action at
the site of rupture of human atheroma. Gamma interferon
can weaken the fibrous cap by inhibiting biosynthesis
of new collagen by the smooth muscle cell, impairing
the ability of the smooth muscle cell to repair
and maintain the plaque's fibrous cap. The level
of collagen in the fibrous cap depends on the rate
of synthesis as well as the rate of breakdown. The
triple collagen fiber is ordinarily a very strong,
biochemically-resistant structure.
Role of matrix metalloproteinase
Only a handful of enzymes are capable
of attacking collagen, notably the interstitial
collagenases. The interstitial collagenases, members
of the matrix metalloproteinase (MMP) family, can
make an initial proteolytic cleavage, breaking the
collagen fiber into three-quarter and one-quarter
fragments. Normal human arteries fortunately do
not contain active forms of interstitial collagenase.
Libby's group showed some years ago
that foam cells derived from both macrophages and
smooth muscle cells in the mature human atherosclerotic
plaque overexpress the collagen- degrading enzyme
MMP-1. However, the presence of immunoreactive MMPs
do not imply biologic activity. MMPs are synthesized
as pro-enzymes that require processing to attain
activity. Antibodies do not distinguish the zymogen
from the active form of the MMP. Potent endogenous
inhibitors of the matrix MMP are widely distributed.
Libby's laboratory has described three of the four
known tissue inhibitors of MMP found in the atheroma.
Recent work by Libby's laboratory
shows there are two enzymes, MMP-1 and MMP-13, expressed
in the atherosclerotic plaque that can break down
collagen, including in situ evidence of collagenolysis
by MMP in the human atherosclerotic plaque. However,
a break in the fibrous cap would not matter but
for the ensuing thrombosis.
What causes the thrombogenicity
of the lipid core?
For more than a decade it has been
known that macrophages in the core of the human
atherosclerotic plaque overexpress tissue factor,
a potent procoagulant. But, until recently the molecular
switch that turns on tissue factor expression was
unknown. The usual soluble cytokines localized in
the atheroma do not induce macrophage tissue factor
expression. T cells can induce macrophage tissue
factor by contact, but the signal remained elusive.
Recent work from Libby's laboratory
likely identifies the missing link between the T
cell and the lymphocyte, which are side by side
when the human atherosclerotic plaque ruptures.
They have studied a new signaling system known as
CD 40 ligand, a surface bound signal. The usual
soluble cytokines do not induce appreciable tissue
factor activity, but recombinant CD 40 ligand and
the membranes of activated T cells in a CD-ligand
dependent manner briskly induce tissue factor gene
expression. The target gene product tissue factor
is expressed in the same cell that gives rise to
the receptor CD 40 that binds CD 40 ligand. Thus,
they believe that CD 40 turns on tissue factor expression
during an inflammatory state in the arterial intima.