Therapeutics
in the 21st Century |
|
The practice of medicine, particularly cardiovascular
medicine, in the 21st century will be
increasingly driven by the improved understanding
of the molecular mechanisms of disease. Nabel illustrated
this with a hypothetical case study in the year
2015 of a male patient presenting for a routine
exam, concerned about his risk for early onset of
colon cancer and coronary artery disease, due to
the early death of his parents from these diseases.
A history and physical show he is in good health,
exercises regularly, has a blood pressure of 110/80
mmHg, heart rate of 65 and a normal cardiovascular
(CV) exam. His cardiac risk factors are male gender
and elevated total cholesterol and LDL.
DNA genotyping is done, as this is very common
in the year 2015, revealing no elevated risk for
colon cancer based on his normal p53 and p16 genes.
A 15% risk for Alzheimer's disease due to beta-amyloid
protein abnormalities is revealed and a 45% increased
risk for coronary artery disease (CAD) as he is
a heterozygote for LDL-receptor mutation or deficiency.
Further testing is done. Exercise tolerance testing
reveals an ST-segment elevation in the anterior
leads at only 5 minutes into testing and a low heart
rate of 122. Magnetic resonance (MR) angiography
is performed, as cineagiography is no longer used,
revealing a proximal LAD lesion. Using real-time
MR, a DNA-coated stent, encoded for an antithrombotic
or antiproliferative gene, is placed in his LAD.
He is then treated with aspirin and a fifth-generation
statin, and a yearly colonoscopy is recommended.
In this invited lecture, Nabel discussed the molecular
mechanisms of some vascular diseases as an entree
to these types of therapeutic approaches, focusing
on human vascular proliferative disease and the
role of cell cycle proteins in regulating vascular
cell proliferation. A novel DNA binding protein
was also reviewed.
|
|
PAGE
TOP
Human vascular
proliferative disease |
|
Atherosclerosis being a systemic disease with complications
occurring at local sites of the circulation led
to the concept of using catheters commonly used
for diagnostics and therapeutics to deliver recombinant
genes into the blood vessel. The pathogenesis of
the atherosclerotic lesion has been well characterized.
In brief, when smooth muscle cells (SMC) are stimulated
by mitogens after vascular injury, the SMC proliferate
within the media of the artery, migrate up through
the internal elastic lamina into the intima where
they continue to proliferate and contribute to the
lesion through the elaboration of the extracellular
matrix as well as proliferation.
Excessive cell proliferation characterizes a number
of human diseases loosely called human vascular
proliferative diseases. These include coronary artery
stenosis, in which stents have treated elastic recoil
but in-stent restenosis remains high. Coronary and
peripheral artery bypass grafts, failure of ateriovenous
fistulas in hemodialysis patients, superficial femoral
artery stenosis, and peripheral artery stenosis
are also vascular diseases characterized by abnormalities
in cell proliferation.
|
|
PAGE
TOP
Cell cycle
regulation of cell proliferation |
|
To elucidate the molecular mechanisms regulating
vascular smooth muscle cell (VSMC) growth in proliferative
disease, Nabel's group has recently focused on
the cell cycle. Since VSMC are plastic they can
re-enter the cell cycle, in contrast to cardiac
myocytes. Normally quiescent with low proliferative
indices in G serum, when VSMC are stimulated to
divide by mitogens they enter the G1 phase of the
cell cycle. Progression through the G1 phase is
regulated by the assembly and phosphorylation of
cyclin and cyclin-dependent kinases, predominantly
cyclin D, CDK-4, and CDK-6, as well as cyclin E
and CDK-2. These are critical for normal progression
through the G-1 checkpoint in the S phase in which
DNA synthesis occurs and there is a commitment to
mitosis. The G1 checkpoint is critical for the phosphorylation
of the retinoblastoma gene product and releasing
E2F and other transcription factors.
Cyclin-dependent kinase inhibitors (CKIs) are endogenous
inhibitors of the cyclin-CDK complexes and two families
have been identified recently. The CIP/KIP family
is comprised of p21, p27, and p57, and the Ink family
is comprised of p15, p16, p18, and p19.
Characterization of the proteins p21 and p27
The protein p27, first identified and cloned by
Toyoshima and Hunter using a yeast two-hybrid approach
using cyclin D as bait, controls cell proliferation
in response to many normal mitogenic stimuli. p27
safeguards against excessive cell proliferation
in specific pathological settings, such as vascular
injury, experimental glomerulonephritis, retinal
dysplasia, and pituitary tumors.
Studies have tried to link the function of p27
with a variety of cancers. Although p27 is not mutated
or deleted within human epithelial tumors, it is
downregulated in breast, colon, and prostate cancer,
likely via enhanced degradation by the ubiquination
system. This occurs by shuttling of p27 from the
nucleus to the cytoplasm where it undergoes ubiquination
by proteosome complexes. This shuttling is mediated
by the Jab1 gene product p38, as recently identified
by Tomoda and colleagues.
p21 is an inducible CKI that alters their activity
in phase G1, and is the human homologue of the FAR1
and SIC1 gene products defined in yeast. It is also
known as WAF1, CIP1, or SDI1, and exists in catalytically
active complexes with PCNA, cyclins, and CDKs. p21
is induced upon DNA damage by p53 and mediates G1
cell cycle arrest, at which point DNA repair is
accomplished before DNA synthesis in the S phase.
In SMC, p21 also has p53 independent effects. p21
null mice exhibit B cell lineage abnormalities and
late onset tumors, in contrast to the p53 null mice
that have early onset tumors.
Elucidating the role of p21 and p27 in vascular
disease
The mitogenic pathways by which growth factor receptors
and signal transduction pathways allow SMC proliferation
has been a large focus of vascular biologists. Less
understood is what turns off cell proliferation
during the later phases of arterial repair.
Nabel and colleagues hypothesized that p21 and
p27 may be important negative regulators of cell
growth. To address this question, they first showed
that p27 is constitutively expressed in SMC within
the media as well as by occasional SMC and endothelial
cells in the intima in an uninjured pig artery.
After balloon catheter injury to the artery, p27
is rapidly downregulated and is at a very low level
until day 7, during the time when cell proliferation
in the lesion is increasing. By day 14, p27 expression
is seen again and increases over time. At day 60
when the lesion is quiescent with low proliferation
indices p27 continues to be constitutively expressed.
In the lower region of the intima, SMC express p27,
procollagen, and TGF-beta.
In contrast, p21 is not constitutively expressed
in the normal artery in this model, and its expression
is only seen in the later phases of arterial repair
at day 21. At day 60 p21 expression is turned off.
Working hypothesis for p21 and p27
The initial working hypothesis, based on these
studies, was that p27 is expressed in a normal quiescent
artery potentially to keep cells out of cycle. However,
upon vessel injury, p27 is rapidly downregulated
and a number of mitogens are produced by SMC, platelets,
and other cells that stimulate cell proliferation.
Proliferation peaks at about one week in this animal
model and then rapidly declines.
Studies of p21 and p27
Expression, cell culture, and in vivo studies of
the CKIs p21 and p27, discussed hereafter, have
shown they are expressed in both human and porcine
tissue, but the overexpression of these lead to
negative growth regulation. The mechanism is likely
mediated through differential regulation of CDK-2.
The CKIs p21 and p27 inactivate cyclin-CDK complexes
in the G1 phase, leading to cell cycle arrest, and
thus function in growth regulation and wound repair.
p27 is constitutively expressed in normal arteries,
is downregulated after arterial injury, becomes
upregulated during the later phases of arterial
repair, and is inversely correlated with VSMC proliferation.
p21 acts in concert with p27 in the later phase
of wound repair.
The KIP/CIP and Ink CKIs differentially regulate
CDK-2 and CDK-4 in VSMC. This leads to differences
in the inhibition of VSMC proliferation in vitro
and in vivo. Expression of p27 and p21 in VSMC inactivated
CDK2 and CDK 4 activity. These different molecular
mechanisms could account for observed differences
in vivo.
Expression Studies of p21 and p27
In the animal model Ross and colleagues defined
an autocrine pathway by which extracellular matrix
secreted by SMC can feed back onto the SMC via the
collagen receptor, alpha-2 beta-1 integrin, to cause
an upregulation of p27 and p21, simultaneous G1
arrest, and downregulation of cyclin E and cyclin
A. Thus, the CKIs are endogenous negative regulators
of SMC that can turn off cell proliferation by promoting
G1 arrest in the later phases of arterial repair.
In coronary arteries of patients who had undergone
cardiac transplantation, Nabel's group found expression
of p27 within SMC in the media and the intima. However,
p21 was not seen in diffuse intimal lesions and
was rarely seen in early atherosclerosis, but was
commonly seen in late atherosclerosis, often in
association with p27. In contrast the Ink CKI, p16,
was not present in any of the coronary lesions,
similar to their findings in the pig model where
p16 was expressed only very early after vascular
injury for unapparent reasons. Expression of p21
and p27 in SMC is also seen in new capillaries formed
within the atherosclerotic plaque, and within macrophages
within the intimal lesion in non-replicating cells.
Cell Culture Studies of p21 and p27
A series of in vitro cell culture studies were
undertaken to understand the mechanism of CKI regulation.
p16, p21, or p27 were expressed in porcine SMC using
adenoviral vectors. Expression of p21 and p27 led
to complete inhibition of cell growth, while p16
led to only partial inhibition.
A series of kinase assays then showed that the
Ink and the CIP/KIP family members inhibited phosphorylation
of cyclin D and CDK-4. Only p21 and p27, in contrast
to p16, inactivated CDK-2 kinase activity in SMC.
The differential regulation of cyclin E and CDK-2
activity in SMC is thought to account for the powerful
effect of the CIP/KIP family members in regulating
SMC growth, as opposed to the Ink family member
p16.
In Vivo Studies of p21 and p27
Using an in vivo animal model of gene transfer,
these observations were studied further. Balloon-injured
pig arteries infected with either a p16 or p27 adenoviral
vector showed that p27 inhibits SMC proliferation.
At 3 weeks there was a reduction in lesion size
by p27 infected arteries, as opposed to p16 or control
arteries treated with either saline or control adenoviral
vector.
The effects seen with p27 mirror the effects previously
seen in response to p21, where overexpression of
p21 also limits the development of the lesion at
3 weeks compared to the two control groups.
|
|
PAGE
TOP
Do p21 and
p27 protect against vascular disease? |
|
A mouse model of vascular injury and one of atherosclerosis
formation are being used to test whether p21 and
p27 are protective against vascular disease. These
studies are ongoing, but preliminary results show
that p27 null mice display abnormalities of growth
regulation. The major phenotype is benign hyperplasia,
predominantly in the endocrine organs (thyroid,
ovaries, adrenal, and pituitary glands). VSMC quiescence
is maintained by p27 and p21. The absence of these
CKIs lead to accelerated VSMC growth and lesion
formation during arterial repair.
Vascular injury studies
To address vascular injury, a p21 knockout, a p27
knockout, and a p21/p27 knockout is being studied
using a C57Bl6 mouse as a control. Each was backcrossed
against the C57Bl6 for 10 generations to remove
any strain differences. The p27 knockout mouse is
larger than the control mouse at an equivalent age
because the absence of p27 leads to benign hyperplasia
of many organs.
The development of a mouse model to test the response
to vascular injury was undertaken. In the femoral
artery wire, injury led to a very brisk lesion within
several weeks. By 3 weeks in the C57Bl6 mouse, there
was a very brisk intimal lesion and by 4 weeks the
vessel was nearly occluded; this is not thrombus
formation as wire injury denudes endothelium but
does not produce thrombus.
The preliminary findings at two weeks post-vascular
injury show a very small intimal lesion forms in
the C57Bl6 mouse, while the largest intimal lesion
is seen in the p27 null mouse. A representative
photomicrograph from the p27 null mouse shows the
SMC have a very dysplastic, unorganized feature
within the lesion, not the concentric array of SMC
interspersed with elastic tissue. This dysplastic
appearance is very characteristic of that seen in
some of the endocrine organs in the p27 null mice.
In contrast, the lesion size in a p21 null mouse
is about one-half that seen in the p27 at two weeks.
However, at 4 weeks the p21 catches up to the p27
null mouse. Interestingly, at 4 weeks in the p27
knockout the cellular proliferation is exuberant
enough to cause a nearly complete occlusion of the
blood vessel. In the combination p21/p27 knockout
there is no difference between the single or the
double knock-out mouse at 2 or 4 weeks.
Atherosclerosis formation studies
To test their hypothesis about atherosclerosis
formation, they crossed the CKI deletion against
an apoE null predisposing the animal towards atherosclerosis
development, and created a series of double and
triple null animals (p21/apoE null, p27/apoE null,
p21/p27/apoE null). The C57Bl6 served as the control.
The absence of apoE accelerates intimal formation.
Immunohistochemistry shows the C57Bl6 lesion is
predominantly an SMC-rich lesion, with macrophages
in the adventitia and a paucity of T cells. In contrast,
in the apoE null mouse, there is a combination of
an SMC-rich and inflammatory-rich lesion, with a
larger number of macrophages within the intima compared
to the C57Bl6, and occasional T cells in the adventitia
and intimal lesion. The studies in the apoE crosses
are underway.
|
|
PAGE
TOP
Novel DNA
Binding Proteins |
|
To understand the regulation of p27 itself within
SMC, Nabel's group used a yeast two-hybrid approach,
with a human B cell library, to potentially clone
novel DNA binding proteins. The carboxy terminal
domain of p27 was used as bait to avoid pulling
out a number of the cyclins and CDKs.
Initially, about 50 clones were assayed based on
the strength of their beta-gal staining. Nabel discussed
the clone initially called C21 and subsequently
named hKIS. C21 bound p27 in the carboxy terminal
domain and had very strong beta-gal staining.
Since C21 is 99% similar to a protein previously
identified in the rat, serine/threonine kinase and
called kinase interacting stathium (KIS), they concluded
that C21 was a human homologue to the rat KIS and
named it hKIS. hKIS is a 46.5 kDA protein consisting
of an NH2 terminal serine/threonine kinase consensus
region and a carboxy terminal region that binds
p27.
Through a series of GST proteins they found that
hKIS specifically bound p27, in contrast to other
CIP/KIP CKIs, p21 and p57, as well as the Ink CKI
p16. A series of biochemical studies have shown
that hKIS functions to phosphorylate p27 in the
N terminal region. A series of mutational studies
showed the phosphorylation site is at position 10
on serine. When serine is substituted for by alanine
at position 10 phosphorylation of p27 is abrogated
by hKIS.
hKIS is located predominantly in the nucleus as
identified by GFP studies, while cellular extracts
show that p27 is located in the cytoplasm and the
nucleus. The binding of hKIS to p27 is predominantly
nuclear.
In SMC, CKI can be expressed alone or in combination
with hKIS to look at the effects on cell cycle.
When hKIS is overexpressed, normal cell cycle progression
is maintained. When p27 is overexpressed, G1 arrest
is produced. However, when p27 and hKIS are co-expressed
while controlling for transfection levels, hKIS
abrogates cell cycle arrest by p27 allowing progression
into G2M. In contrast, p21 alone arrests the cell
cycle and overexpression of p21 and hKIS continues
cell cycle arrest.
hKIS working hypothesis
Nabel's group hypothesizes that hKIS is a binding
protein that uniquely regulates p27 function. hKIS
binds p27 through the carboxy terminal domain and
then phosphorylates and inactivates p27 upstream
of a cyclin-CDK binding site.
Further, they hypothesize that hKIS may be an upstream
regulator of p38, important for inactivating p27,
and preparing it for translocation out of the nucleus
into the cytoplasm and degradation. As a regulatory
protein for p27, hKIS can bind and thus inactivate
p27 and potentially prepare it for binding by the
Jab 1 gene product, p38. p38 has been identified
to be important for translocation of p27 from the
nucleus out to the cytoplasm where it is then degraded
by ubiquitin-conjugation system within proteosomes.
The studies with hKIS are ongoing.
|
|
PAGE
TOP
Molecular
regulation of proliferation and potential therapeutic
applications |
|
Several approaches may be taken to investigate
the methodologies for vascular gene transfer to
develop approaches for treating human vascular proliferative
diseases. Nabel's group has predominantly focused
on regulators of the cell cycle. Other investigators
have pursued other approaches, including the overexpression
of a mutant form of the Rb gene product by Leiden
and transcription decoys to E2F by Dzau.
Nabel's group used a thymidine kinase (TK) approach
early on and she reviewed these studies as proof
of principle that SMC proliferation could be disrupted
during vascular injury and translated into therapeutic
application.
The rationale for using TK is that it can achieve
a bystander effect in cells, and hence create a
larger biological effect in a greater number of
cells than the actual number of cells transfected.
This was important when the studies were begun five
years ago because the efficiency of gene transfer,
even with adenoviral vectors, was not overwhelming
in the vasculature.
Normally TK is not expressed in mammalian cells.
When it is overexpressed using a vector approach
it encodes for an enzyme that phosphorylates the
prodrug ganciclovir (GCV) or acyclovir (ACV), nucleoside
analogues, into a phosphorylated form that is incorporated
into replicating DNA, causing chain termination
and cell death. By a poorly described bystander
effect, byproducts are then diffusable to adjacent
cells where again they are incorporated into replicating
DNA, and can kill adjacent dividing cells in which
the gene is not expressed.
The TK approach has now been tested in a number
of animal models of restenosis and vascular proliferative
disease by a number of laboratories in the US with
similar findings across groups. It is a fairly potent
way to disrupt VSMC proliferation. Using heterologous
promoters is non-permissive in that they will also
disrupt proliferation of other cell types. Nabel
showed in rabbit hyperlipidemic arteries, for example,
that macrophage proliferation is inhibited.
Modifying the TK vector for human therapy
Nabel's group studied whether they could modify
and enhance the TK vector before attempting potential
human therapy. They incorporated an internal ribosomal
entry site to obtain expression of two genes from
the vector. Guanylate kinase (GK) was used to enhance
phosphorylation of TK. The TKciteGK vector was then
tested with a variety of promoters in an adenoviral
construct to test the strength of the heterologous
promoters, RSV, EF1-alpha, and CMV in SMC. The two
pro-drugs ACV and GCV were also tested.
In testing SMC in culture, the largest percentage
of cells were killed with the combination of the
adenoviral vector with CMV and GCV. This was then
used in an animal model of vascular proliferation
using a channel balloon with a stent mounted on
the balloon catheter. When the catheter is inserted
within the artery, the stent is deployed, and the
vector can be infused through the balloon catheter
through very small pores within the channel of the
balloon. The vector is then mechanically introduced
into the stretched artery. Thus, two components
that lead to the pathophysiology of restenosis is
treated: elastic recoil through stent deployment
and the proliferative component through the vector
TKciteGK. A number of other anti-proliferative vectors
could be used.
Nabel's group has tested this approach in peripheral
pig arteries using an EF1-alpha and a CMV promoter
and two different pro-drugs, ACV and GCV. Saline
controls were used. The smallest intimal lesions
in the artery after 3 weeks were obtained with the
combination of the CMV promoter with the TKciteGK
vector in combination with GCV.
This seemingly straightforward and basic pharmacology
is the type of testing of promoter-vector-drug delivery
devices that will be absolutely essentially as these
technologies are brought forward to the clinical
arena.
|
|
PAGE
TOP
Is cell specific
regulation of gene expression possible? |
|
Collaborating with another laboratory, Nabel has
recently looked at a SMC-specific promoter, SM22-alpha,
which was used to drive expression of two different
reporter genes as well as the TKciteGK vector. These
vectors have been tested in a series of cell culture
and animal models.
With the SM22-alpha promoter in cell culture there
is faithful expression in pig aortic SMC, pig jugular
venous SMC, and mouse aortic SMC. But, not in non-SMC
lines in NIH3T3 cells or pig endothelial cells.
In contrast, the heterologous promoter RSV drives
expression of the reporter gene in all cell types.
The expression being driven by an SM22-alpha promoter,
in arterial SMC at least, is at least 3 log-fold
lower than by an RSV or a CMV promoter. This suggests
that the levels of expression achievable in vivo
would be far less with a cell specific promoter.
The cell specific promoter might offer increased
safety advantages for in vivo arterial applications.
|
|
PAGE TOP
In vivo model
of SMC-specific promoters |
|
In a pig balloon injured-femoral
artery overexpressing the vectors 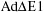 ,
AdSM22-alpha-hpAP, AdRSV-hpAP, at 5 days the reporter
gene is driven by the RSV promoter and there is expression
by SMC in the upper regions of the media, the subintima,
and by endothelial cells. In contrast the SM22-alpha
promoter drives exclusive expression of SMC in the
media, the lower regions of the intima and by endothelial
cells. Thus, expression with a SMC-specific promoter
is faithful in vivo, although the levels of expression
are much lower. These are the types of approaches
required to bring these molecular concepts forward
into safe treatments for human vascular disease. |
|
PAGE
TOP
|
In patients with cardiovascular disorders, it will
be increasingly important to understand and apply
a molecular understanding of the disease for both
diagnostics and therapeutics.
This requires a very good understanding of the
genetic structure and function of the different
proteins that regulate cell growth and cell differentiation.
It is possible to dissect out molecular mechanisms
of vascular disease using expression studies and
knockout studies in mice, and these can used as
a basis for developing genetic therapies.
A word of caution was delivered by Nabel. As researchers
proceed with these approaches, it is important to
focus not only on doing good science in the sense
of having a very good understanding of the therapeutic
genes, but also focusing on understanding the safety
and potential toxicity of the vectors and the delivery
devices. It is essential to take the time necessary
to go forward with these studies in a very careful
and logical manner. This might require a few more
years to achieve the goal, but in the final analysis
this will benefit the research and, with time, patients.
|
|
PAGE
TOP
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2000
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
