| In their efforts to understand
the basic mechanisms for gene expression during the
transition from a normal heart to hypertrophic growth
to heart failure, Olson and colleagues have identified
two signal transduction pathways involved in hypertrophic
growth. Both are mediated by a calcium calmodulin-dependent
enzyme, calcineurin and CaM kinase, which are thought
to be sensors for hypertrophic stimuli and act through
very different cell biological mechanisms. Calcineurin
acts by dephosphorylating a transcription factor that
enables it to go to the nucleus where it engages a partner
protein to switch on hypertrophy. CaM kinase acts by
phosphorylating a corepressor that causes its export
to the cytoplasm, which then releases MEF-2 to switch
on its downstream target changes. Perturbing this step
can block hypertrophy in response to diverse signals,
including those known to act through calcineurin activationindicating
there is crosstalk between these two pathways, particularly
at the final common step in the nucleus. |
|
Calcineurin-Mediated
Pathway |
In this hypertrophic signaling
pathway, cell surface agonists or activity of cardiomyocytes,
known to elevate intracellular calcium, can activate
a cytoplasmically-located protein phosphatase called
calcineurin. It is believed that calcineurin functions
as an intracellular sensor for calcium signaling and
that it transduces hypertrophic signals downstream through
a recipient transcription factor known NF-AT. NF-AT
is phosphorylated in the cytoplasm of cardiomyocytes,
but when calcineurin is activated it becomes dephosphorylated.
Upon dephosphorylation of NF-AT, this transcription
factor is enabled to translocate into the nucleus where
it interacts with a cardiac-restricted zinc finger transcription
factor, GATA-4. This protein-protein interaction establishes
a unique transcriptional code that switches on the gene
regulatory program for cardiomyocyte hypertrophy and
cardiac growth.
In subsequent experiments they showed that activating
the calcium calmodulin-dependent phosphatase calcineurin
or simply dephosphorylating NF-AT is sufficient to evoke
the full hypertrophic response in vivo that progresses
to dilated cardiomyopathy and ultimately to sudden cardiac
death.
Calcineurin phosphatase can be inhibited by a family
of drugs, one of which is cyclosporine A (CsA). Thus,
this pathway for cardiac hypertrophy generated a great
deal of interest. Olson and colleagues showed that treatment
with CsA in calcineurin-transgenic mice maintained their
heart size to that similar in the wild-type mice, whereas
the non-treated transgenic mice developed dilated cardiomyopathy
(DCM) and sudden death. CsA also prevented activation
of the fetal gene program, blocks fibrosis, and prevents
sudden death in the transgenic mice. The majority of
mice models tested by other laboratories could be rescued
from hypertrophy by CsA treatment, including models
that developed hypertrophy in response to myocardial
infarction (MI), salt-induced hypertension, G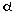 q
activation and sarcomeric dysfunction. Thus, a diverse
array of stimuli known to elevate intracellular calcium
seem to activate hypertrophy through an obligatory downstream
step involving calcineurin activation. q
activation and sarcomeric dysfunction. Thus, a diverse
array of stimuli known to elevate intracellular calcium
seem to activate hypertrophy through an obligatory downstream
step involving calcineurin activation.
To confirm the role of calcineurin in cardiac hypertrophy,
a genetic model in which calcineurin was inhibited by
the muscle calcineurin inhibitory protein (MCIP), which
is expressed in a muscle-specific manner with the highest
expression in the heart and in skeletal muscle has been
used by Olson and colleagues and other investigators.
They have shown that MCIP is also sufficient to prevent
hypertrophy in response to a diverse array of stimuli
with no apparent consequence on the overall health or
endurance of the animal. Further study revealed that
MCIP is upregulated from normal hearts to hypertrophic
hearts and it is also upregulated from hypertrophic
to failing hearts in response to calcineurin activation
and was a common response to diverse types of hypertrophic
signals.
The simplified model developed based on these studies
for the calcineurin signaling pathway activation and
regulation in cardiomyocytes is that calcium signaling
activates the phosphatase, which dephosphorylates NF-AT,
enabling it to translocate to the nucleus. The MCIP
gene seems to be one of the most sensitive genes in
cardiomyocytes to calcineurin signaling, and it has
been that there are 15 tandem NF-AT binding sites in
the promoter of the MCIP gene. MCIP expression causes
it to feed back on calcineurin, binding directly to
the catalytic subunit and diminishes calcineurin activation.
It is thought that this is a built-in response by the
heart to prevent further hypertrophic stimulation. |
PAGE
TOP
|
CaM Kinase
Signaling Pathway |
Calcium calmodulin-dependent protein (CaM) kinase
has been shown to be elevated in biopsy samples from
heart failure patients. Inhibitors of CaM kinase have
been shown to interfere with hypertrophic signaling
in cultured myocytes. Thus, the role of CaM kinase in
hypertrophic signaling was explored by Olson and colleagues.
A family of developmentally important transcription
factors known as the MEF2 family serves as the sensor
for a diverse set of hypertrophic signals. MEF-2 proteins
bind to fetal cardiac genes and bind to a host of stress-inducible
genes up-regulated during cardiac hypertrophy. MEF-2
proteins function as partner proteins for other lineage-restricted
transcription factors. It is known that these proteins
dimerize with members of the MyoD family and this protein-protein
interaction establishes a code that selectively activates
every downstream event in the skeletal muscle pathway.
MEF-2 factors are essential for cardiomyocyte differentiation,
as shown by genetic mutations in fruit flies and in
mice. These MEF-2 factors bind directly to the promoters
of downstream fetal cardiac genes and are required for
ventricular development. MEF-2 proteins regulate vascular
development by switching on differentiation genes and
controlling morphogenesis of the vasculature.
MEF-2 sensor mice were developed by Olson and colleagues
to enable the study of the transcriptional activity
of MEF-2 proteins in vivo in the adult heart. Surprisingly,
they found that the developmental control protein MEF-2
is present in the adult heart in an inactive form. Further
study showed that the MEF-2 protein is a sensor of hypertrophic
signals evoked by CaM kinase activation in the adult
heart, and that these signals switch the protein from
an inactive to active state. Calcineurin signaling can
also activate the MEF-2 transcription factor.
Olson and colleagues then sought to determine how this
protein residing in the nucleus of a cardiomyocyte can
sense the activation of signal transduction pathways
in the cytoplasm and how the activation of this transcription
facto can reprogram a cardiomyocyte to switch on the
entire hypertrophic gene regulatory program. They found
that MEF-2 interacts with a family of chromatin remodeling
enzymes, known as histone deacetylases (HDAC), shown
to play a central role in regulating changes in gene
expression in response to growth, development and cell
signaling.
HDACs inhibit transcription by removing acetate groups
from histones, causing chromatin condensation and transcriptional
repression. The finding that a family of transcriptional
repressors, HDACs, could interact with MEF-2 was exciting
since it was known that MEF-2 in the adult heart was
transcriptionally inactive and that HDACs were highly
expressed in the adult heart. Further study showed that
the association of MEF-2 with HDAC represses the ability
of MEF-2 to activate its targets. Activation of CaM
kinase overcomes this repression, allowing MEF-2 to
go from complete inactivation to super-activation. Further,
they showed that MEF-2 simply serves as a platform for
recruitment of a corepressor. In an adult cardiomyocyte,
MEF-2 binds DNA, is a complex with HDAC5, and hypertrophic
genes are silent. CaM kinase signaling disrupts this
complex and causes the export of HDAC5 from the nucleus
to the cytoplasm.
Extensive biochemical experiments by Olson and colleagues
showed that HDAC5 contains two direct phosphorylation
sites for CaM kinase signaling. These two sites are
the molecular targets through which CaM kinase signals
to the MEF-2-HDAC complex. HDAC5 is nuclear until CaM
kinase is active and then it is exported to the cytoplasm.
MEF-2 remains in the nucleus. If those two sites are
mutated so they cannot be phosphorylated, HDAC5 irreversibly
locks onto MEF-2 and it cannot be activated.
Phosphorylation of these two sites in HDAC5 has two
consequences. First, it recruits 14-3-3 proteins (phosphorylation-dependent
chaperone proteins) to both sites. The interaction of
the two sites and 14-3-3 is thought to induce a conformational
change in HDAC5 that pries it away from MEF-2. Second,
after 14-3-3 binds to these two sites, the protein-protein
complex is exported to the cytoplasm. It is thought
that this occurs because 14-3-3 binding masks the nuclear
localization sequence of HDAC5 that is located directly
between these two sites. Further study by Olson and
colleagues showed that HDAC5 is the brake that blocks
hypertrophy and that eliminating this repressor will
cause cells to spontaneously hypertrophy in the absence
of any upstream signaling molecule in the hypertrophic
pathway.
Through further studies, Olson and colleagues have concluded
that these two sites are the final common pathway through
which multiple signals flow. These are signals elicited
by binding of endothelin to the cell surface receptor,
binding of adrenergic agonists and activation of pathways
induced by fetal bovine serum. By perturbing a final
common pathway, it may be possible to interfere with
cardiac hypertrophy both in vitro and in vivo. |
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2001
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
