 |
|
|
|
| Heart Failure as a Maladaptive
Consequence of Cardiac Hypertrophy |
|
Shigetake Sasayama, M.D.,
Ph.D.
Kyoto University
Kyoto, Japan |
|
 |
|
|
|
|
|
|
| The maladaptive consequences
of cardiac hypertrophy constitute important underlying
mechanisms of the syndrome of heart failure. This lecture
reviewed the body of animal and clinical research of
adaptive and maladaptive hypertrophy conducted by Sasayama
and colleagues throughout his career and related the
findings with new treatment approaches. |
|
Physiological
and Structural Changes |
|
The left ventricle (LV) can augment its performance
during chronic volume overload with no further use
of the Frank-Starling mechanisms, and hypertrophy
is the primary adaptive mechanism to maintain wall
stress within a certain limit. These findings are
supported by serial studies with left ventriculography
that revealed progressive dilation of the LV chamber
and a moderate increase in the LV wall thickness during
chronic volume overload in the canine model. The mean
velocity of the circumferential shortening showed
no appreciable change, and the wall stress was elevated
markedly in the early stage but gradually decreased
with the development of hypertrophy. Serial studies
in the canine model also indicated that hypertrophy
does not necessarily result in depressed contractility.
In the clinical setting, it was shown that the moderately
hypertrophied ventricle exhibits hyperfunction as
a pump, but does not have intrinsic depression of
cardiac contractility, while the contractility of
the ventricle with advanced hypertrophy was substantially
decreased.
The sequence of events during the development of
hypertrophy comprises three stages. Stage one is myocardial
damage and impairment of contractility; two, stable
hyperfunction, in which normal myocardial function
is restored; three, gradual deterioration of myocardial
function leading to overt heart failure.
|
PAGE
TOP
|
Inciting
Events in Development of Heart Failure |
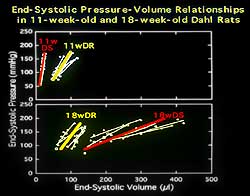 |
| Figure
1. The end systolic pressure volume relations
changes are consistent with substantial contractile
depression. |
| Click
to enlarge |
|
No consensus
exists regarding the factors responsible for the transition
from compensated hypertrophy to heart failure, despite
repeated descriptions of decompensated hypertrophy.
Sasayama and colleagues developed an experimental rat
model that permitted the evaluation of the sequence
of ventricular responses to a sustained increase in
workload. The changes in the end systolic pressure volume
relations, shown in Figure 1, were consistent with substantial
contractile depression. |
|
The importance of understanding inciting events rather
than the terminal results is supported by the findings
from studies with this experimental rat model, as
the changes en route to heart failure occurred in
a compensatory manner as early as 11 weeks. The initial
event is the influx of calcium into the myocytes through
the sarcolemmal calcium channels, triggering the release
of the activator calcium from the local pool. In the
failing stage, there was a definite ventricular dysfunction
with a reduced peak level of tension development with
a marked delay in the tension decay. These changes
were associated with corresponding changes in the
calcium transient. Impaired contractile response to
beta adrenergic stimulation in the failing heart was
primarily assigned to a decreased number of beta receptors
and an increase in inhibitory G1 protein in the failing
heart compared to the control animals; changes that
occurred in the compensatory stage.
Elevated circulating levels of cytokines have been
noted in patients with chronic heart failure. A growing
body of evidence shows that a major subset of heart
disease may be expressed via nonlethal alteration
in myocyte function induced by immune cells and their
cytokines. Data from Sasayama and colleagues show
that plasma TNF-alpha levels are elevated in a large
percentage of patients with acute myocarditis and
dilated and hypertrophic cardiomyopathies, and that
in patients with dilated cardiomyopathy cytokines
such as IL-beta, IL-2, interferon gamma, and TNF-alpha
were significantly upregulated.
|
PAGE
TOP
|
Myocarditis
Precipitates Dilated Cardiomyopathy |
| Dilated cardiomyopathy (DCM)
constitutes the most common cause of heart failure,
and it has long been postulated that myocarditis may
precipitate DCM. Work by Sasayama and colleagues showed
in a murine model that three months after myocarditis
virus infection that heart rate increased with further
dilation of ventricular chamber, and hypertrophy and
interstitial fibrosis developed in the absence of an
acute inflammatory process. The heart assumed the characteristic
pattern of DCM, and the failing heart was characterized
by a downward shift of the end systolic pressure volume
relation and diastolic dysfunction with an upward shift
of the diastolic pressure volume relations. Il-1 beta
and TNF-alpha were upregulated within three days of
infection, whereas serine infiltration was not apparent.
Il-2 and interferon gamma then became detectable with
a substantial increase in serine infiltration. Expression
of all of the cytokines peaked on day 7 but persisted
for 80 days, even when the infectious virus was not
longer serologically detectable. Further study showed
that the cytokines initiate beneficial cell-protective
immune responses in the acute phase, but the sustained
induction of cytokines is detrimental and produces destructive
immune responses directed against the myocardium. |
PAGE
TOP
|
Proinflammatory
Cytokines |
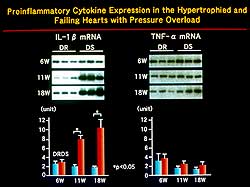 |
| Figure
2. In animal studies, mechanical stress resulted
in gene expression of cytokines in stretched endothelial
cells compared to static control cells. |
| Click
to enlarge |
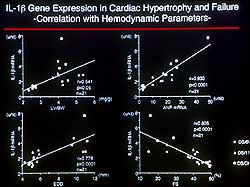 |
| Figure
3. A positive correlation was seen between IL-1
beta mRNA and ventricular mass, chamber size,
and ANP mRNA and an inverse relation was seen
with fractional shortening. |
| Click
to enlarge |
|
Mechanical stress
was shown in animal studies to result in gene expression
of cytokines. Figure 2 shows that IL-8 and MCP-1 were
significantly upregulated in stretched endothelial cells
compared to static control cells. MCP-1 mRNA was augmented
in the compensated hypertrophic rat at 11weeks and further
upregulated in failing hearts at 18 weeks. MCP-1 is
a member of the chemokine family that activates T lymphocytes.
The recruitment of monocytes and macrophages by these
chemokines may be a pivotal step for the expression
of cytokines. The amount of IL-1 beta mRNA showed a
clear positive correlation with ventricular mass, chamber
size, and ANP mRNA and was inversely related with fractional
shortening (Figure 3). IL-1 beta caused myocyte growth
with re-expression of fetal genes and importantly modified
the remodeling process and ventricular function. Studies
revealed that a cytokine inhibitor specific for IL-1
beta significantly improved survival in a rat model
of pressure-overload hypertrophy. |
PAGE
TOP
|
RAS and Transition
to Heart Failure |
| The renin angiotensin system
(RAS), including angiotensin II, angiotensinogen, and
ACE inhibitors, were upregulated at 11 weeks in the
rat model of pressure overload hypertrophy—findings
that are consistent with the hypothesis that locally-produced
angiotensin II may act as an endogenous growth factor
in the myocardium. RAS activation remains at the same
level in the failing heart, this angiotensin II does
not appear to be a critical factor mediating the transition
to heart failure. |
PAGE
TOP
|
|
|
| Figure
4. Endothelin-1 (ET-1) concentrations were markedly
increased as the heart transitioned from hypertrophy
to heart failure. |
| |
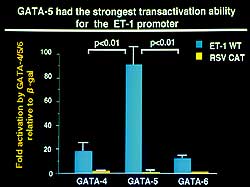 |
| Figure
5. GATA-5 expression was markedly elevated in
the failing heart showing it to have the strongest
transactivation ability for the ET-1 promoter
in animal studies. |
| Click
to enlarge |
 |
| Figure
6. Functional and hemodynamic parameters were
significantly improved with bosentan and temocapril.
Bosentan had no effect on ventricular mass in
contrast to temocapril. |
| Click
to enlarge |
|
Serum and myocardial concentrations of endothelin-1
(ET-1) were markedly increased four-fold and five-fold,
respectively, during the transition from compensated
hypertrophy to heart failure in the pressure overload
hypertrophic model (Figure 4). Thus, myocardial synthesis
of ET-1 was considered to play a significant role
in the transition from compensated hypertrophy to
heart failure. Local synthesis of ET-1 was found to
be mediated by acetylated conversion from big ET-1
to ET-1 in the rat cardiac myocyte.
Endothelin converting enzyme (ECE-1) mRNA was not
affected in compensated hypertrophy, but was significantly
upregulated in the failing heart. ECE-1 was therefore
considered to play a significant role in increasing
the conversion from big ET-1 to ET-1 during the development
of heart failure. Experiments of the transcriptional
activity of ET-1 promoter in the upregulated expression
of ET-1 in the myocardial cells showed that mutation
of the GATA motif in the ET-1 promoter reduced the
basal transcriptional activity by half and completely
abolished the phenylephrine-mediated increase in transcription.
Thus, GATA was considered essential for full transcriptional
activity of ET-1. GATA-5 was shown to have the strongest
transactivation ability for the ET-1 promoter (Figure
5), and its expression was markedly elevated in the
failing heart. GATA-5 was thus considered to importantly
relate to intranuclear signal transduction of ET-1
expression in the myocardial cells and the development
of heart failure.
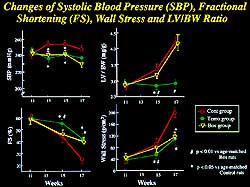
A significant improvement in survival was seen in
the compensated hypertrophic rat with an ET receptor
antagonist (bosentan) and with an ACE inhibitor (temocapril),
compared to complete mortality of the entire control
group by 19 weeks. Bosentan and temocapril significantly
improved the functional and hemodynamic parameters,
but bosentan, in contrast to temocapril, did not affect
ventricular mass (Figure 6). The beneficial effects
the ET receptor antagonist was considered to be mediated
exclusively by improvement of ventricular dysfunction,
while the effect of the ACE inhibitor was related
to the improvement in remodeling.
|
PAGE
TOP
|
| Mast cells are increasingly
becoming of interest in terms of the cytokine-mediated
inflammatory process and response. Mast cells are multifunctional
that contain various mediators, such as histamine, protease,
or leukotriene. Work from Italy shows that myocardial
mast cell density is significantly higher in the failing
heart with cardiomyopathy, and that the mast cell count
can be reduced with an ACE inhibitor. Clear correlations
between the number of mast cells and cellular fractions
were seen. Therefore, mast cells are considered to play
a significant role in the remodeling process and the
progression of heart failure. Studies showed that mast
cell granules decreased the survival of rat cardiac
myocytes in a concentration-dependent manner. Electron
microscopy revealed several membrane-bound cellular
fractions of various sizes containing cytoplasm and
structure of well-preserved organelles—indicating
that mast cells cause myocyte death by apoptosis. Mast
cells are considered to be importantly related to the
progression of heart failure by producing systolic dysfunction
with loss of myocytes by apoptosis and diastolic dysfunction
with proliferation of fibroblasts. |
PAGE
TOP
|
Path from
Hypertrophy to Heart Failure |
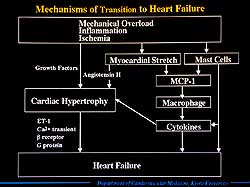 |
| Figure
7. The mechanisms known to be involved in the
transition from hypertrophy to heart failure. |
| Click
to enlarge |
|
The known mechanisms
involved in the transition from hypertrophy to heart
failure are summarized in Figure 7. Briefly, mechanical
overload or inflammatory insult activates various second
messenger systems and induces various oncogenes. Myocardial
stretch also activates the autocrine release of angiotensin
II, which synergistically activates an intracellular
protein kinase cascade and induces various growth factors.
Cardiac hypertrophy then develops, which undergoes various
physiological or biochemical changes, including calcium
degrading protein, calcium transient, and local expression
of ET-1. These changes result in contractile depression
and finally overt heart failure. These pathophysiologic
stages increase the mast cell density in the myocardium,
and myocardial stretch and mast cells induce the chemotactic
factors for macrophages. The recruited macrophages may
be a major source of cytokines, which accelerate myocardial
growth and remodeling and are responsible for reduced
myocyte function. Mast cells also induce cytokines and
are directly related to the development of heart failure. |
| The traditional concept of the length-tension relationship
has been shown to be more complex through animal studies
by Sasayama and colleagues and other investigators.
The modern therapeutic approach to heart failure may
focus on eradicating the maladaptive signaling identified
in these studies and focus on cell targets. The redundancy
of the signaling pathways makes it difficult to completely
inhibit a maladaptive response by a single measure.
|
PAGE
TOP
|
Report
Index | Previous Report | Next
Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2001
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|