| Signal transduction pathways
are the molecular cascades responsible for communicating
information from the exterior to the interior of the
cell. Abnormalities in these pathways play a pivotal
role in the production of heart failure. |
|
| The biochemical paradigm
recognizing that the activation of the neurohormonal
pathways, in particular the renin angiotensin and sympathetic
nervous systems, may importantly contribute to the evolution
of heart failure has supplanted the traditional hemodynamic
paradigm of heart failure. This biochemical paradigm
serves as a basis for current cardiovascular therapeutics
in heart failure. The understanding in the last decade
that abnormalities in myocardial and vascular cell types
causally contribute to heart failure has provided for
new therapeutic opportunities. Understanding molecular
and cell biology and how they contribute to the onset
of heart failure will serve as a platform for new and
effective therapeutic modalities. |
PAGE
TOP
|
Mechanisms
for Compensated Hypertrophy |
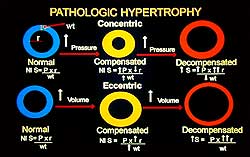 |
| Figure
1. Overload-induced hypertrophy results in a concentric
increase in cardiac contractile units. |
| Click
to enlarge |
|
Hypertrophy,
an increase in cardiac contractile units in a concentric
fashion as a consequence of pressure overload or volume
overload is illustrated in Figure 1. The increase in
contractile units normalizes wall stress, maintains
left ventricular systolic function and maintains cardiac
output. The pressure or volume overload, if sufficiently
intense or prolonged, results in a large, dilated heart
with an increase in wall stress and a mechanical disadvantage
for both pumping and filling.
Abnormalities in signal transduction and how they may
contribute to or cause maladaptive hypertrophy and heart
failure is the focus of much research. Some of the signal
transduction pathways identified as being important
in the production of pathologic hypertrophy and heart
failure, include cytokines, conventional growth factors,
angiotensin, extracellular stresses and mechanical deformation.
For example, if GP130 in the JAK-STAT pathway, which
has been shown to be anti-apoptotic or protective for
normal growth, is knocked out and superimposed with
pressure overload, dilated cardiomyopathy occurs in
the genetically-engineered mouse on the basis of massive
apoptosis. |
PAGE
TOP
|
The protein kinase C (PKC)
pathway seems to occupy a nodal position and can cross-activate
the classical MAP kinase pathway. In other cell types
it has been shown to be important in the production
of pathologic growth and abnormalities of function.
Angiotensin II, endothelin-1, phenylephrine, prostaglandin-F2-alpha
and mechanical deformation can activate protein kinase
C. All are involved in the pathologic development of
pressure overload, volume overload and heart failure.
The coupling of angiotensin and the other hormones to
their cognate receptor activates, by virtue of dissociation
of the Gq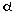 subunit of the heterotrimeric G-protein, phospholipase
C-beta-1 (PLC-ß1). PLC-ß1 acts upon phosphitol
inositol bisphosphate to produce diacylglycerol (DAG),
a general stimulant of PKC, and inositol triphosphate,
a modulator of intracellular calcium homeostasis.
subunit of the heterotrimeric G-protein, phospholipase
C-beta-1 (PLC-ß1). PLC-ß1 acts upon phosphitol
inositol bisphosphate to produce diacylglycerol (DAG),
a general stimulant of PKC, and inositol triphosphate,
a modulator of intracellular calcium homeostasis.
Certain PKC isoforms are calcium-dependent, thus there
is a positive feedback loop for PKC activation of conventional
isoforms. This calcium release may, in incompletely
defined ways, stimulate the activation of phosphatases
and other calcium-dependent kinases.
The different isoforms of PKC include the serine threonine
kinases, initially described as a pathway activated
by partial lipid hydrolysis. The classical protein kinases,
represented by alpha, beta-1, beta-2 and gamma, have
a calcium-binding domain and can be activated by diacylglycerol,
phorbol ester, phosphatidylserine or calcium. The so-called
novel protein kinases, represented by delta and epsilon,
lack the calcium-binding domain and can be activated
independently of calcium by phosphatidylserine and diacylglycerol.
The atypical protein kinases family lacks the diacylglycerol
PMA binding domain and can be activated by phosphatidylserine,
independent of calcium or diacylglycerol.
The existence of different isoforms of PKC has suggested
that they may have different functions, and this fundamental
question is of great research interest. Walsh and colleagues,
using pressure overload in a guinea pig model, showed
that four weeks of descending thoracic aortic banding
was associated with an increase in left ventricular
(LV) mass, which was associated with normal isovolumic
LV contractile function and normal isolated cardiomyocyte
mechanics. But, after 8 weeks of banding a decrease
in isovolumic LV performance and cardiomyocyte function
was observed. Thus, this served as a convenient reagent
to study molecular events common to either the compensated
or decompensated pressure overload hypertrophy and heart
failure.
They also demonstrated that acute mechanical stretch
to a minimum diastolic pressure of 25 mm Hg was sufficient
to activate phospholipase C with inositol triphosphate
accumulation, PKC translocation and activation, mediated
in part by angiotensin II. Further, PKC promiscuous
activation by a phorbol ester depresses isolated LV
mechanics, suggesting that hyperactivation of the PKC
pathway might contribute to contractile dysfunction
in heart failure. |
PAGE
TOP
|
PKC Activation
and Heart Failure |
In an experiment to assess
whether and to what extent LV performance seen as a
clinical consequence of ACE inhibition was due to blockade
of PKC activation, they showed there was no change in
the components of the PKC signal transduction pathway.
But, there was hyperactivation of PKC isoforms and elevated
PKC activity with chronic pressure overload and heart
failure in this guinea pig model. The differential PKC
activation was mediated by increases in Gaq and PLC-ß1
hydrolysis, rather than up-regulation of their expression.
They have also shown that survival in response to LV
pressure overload by descending thoracic aortic banding
was enhanced by ACE inhibition in this guinea pig model
of decompensated congestive heart failure. They demonstrated
that LV hypertrophy and heart failure was diminished
and that LV and cardiomyocyte function was improved,
indicating improvement in LV performance was partly
intrinsic. There was upregulation of the calcium cycling
proteins and an increase in the calcium transient associated
with a decrease in PKC translocation. PKC has been shown
in vitro, by other laboratories, to downregulate the
calcium cycling proteins shown to be dysfunctional and
decreased their abundance in heart failure. |
PAGE
TOP
|
Mechanisms
for PKC Activation |
| Mechanical deformation,
G-protein coupled receptor activation by angiotensin
and endothelin, has been shown to activate PKC. Walsh
and colleagues have shown that angiotensin II, hypoxia
and ischemia activate a variety of the PKC isoforms
via phospholipase-C and tyrosine kinase coupled pathways.
Interestingly, however, oxidative stress using hydrogen
peroxide was able to activate PKC independently of PLC-ß1
or tyrosine kinase signal transduction—which has
important therapeutic implications. |
PAGE
TOP
|
PKC in Pathologic
Hypertrophy |
Walsh and colleagues have
used genetically-engineered mice to understand and characterize
the phenotypic consequences of the genetic modifications
in pathologic hypertrophy. Their work sought to answer
these questions: Is the phenotype due to altered expression
of the transgene itself? Are similar alterations in
protein abundance found in experimental animal systems
or human disease? Is the observed phenotype a consequence
of an insertional effect of the transgene? Is there
ectopic expression of the transgene in a location it
usually does not reside? What are the consequences of
the poison peptide, the nonspecific overexpression in
large non-physiologic, non-pathologic ways of a protein?
How much of the phenotype is due to phenotypic changes?
Walsh and colleagues showed that overexpression of PKC-beta-2
in the transgenic mouse produces ventricular hypertrophy,
myocardial fibrosis, and depression of LV function.
Further, they showed that isoform inhibition, using
the PKC-ß2 inhibitor, prevented or regressed pathologic
hypertrophy and normalized LV function. In terms of
the molecular mechanism for the dysfunction due to overexpression
of PKC-beta-2, they showed a decrease in the rate of
shortening and the rate of re-lengthening, associated
with normal calcium transients. This suggests that the
abnormal in vivo function was, at least in part, due
to a decrease in calcium sensitivity at the level of
the cardiomyocyte. Superfusion with the PKC inhibitor
resulted in relative normalization of isolated LV cardiomyocyte
mechanics.
They concluded that PKC-beta-2 overexpression depressed
myocyte shortening and re-lengthening and is associated
with normal intracellular calcium transients. Using
a survey of phosphorylation targets in the cardiomyocyte,
they found increased phosphorylation of troponin-I,
which resulted in the decrease in myofilament calcium
sensitivity. The PKC-ß2 inhibitor improved cardiomyocyte
function. PKC inhibition might prove to be a useful
therapy for human heart failure.
In another study they showed that PKC isoform expression
and activity are increased in the failing human heart
due to increased protein expression of beta-1, beta-2,
and alpha (calcium-dependent isoforms). There was a
non-significant reduction in the abundance and translocation
of PKC-epsilon. The increased isoform expression was
at least in part due to increases in activity in the
cardiomyocyte that were transcriptionally mediated.
PKC activity was reduced by 45% with the PKC-b2 inhibitor,
again suggesting the importance of PKC activation of
classical isoforms in cardiomyopathic congestive heart
failure. |
 |
| Figure
3. Conceptual framework of the transition from
cardiomyocyte hypertrophy to heart failure. |
| Click
to enlarge |
|
Walsh and colleagues
then went on to show that targeted transgenic overexpression
of the PKC-epsilon isoform produces concentric LV hypertrophy
with no cardiac fibrosis. In vivo LV function was normal
with markedly depressed calcium transients, suggesting
an enhancement of calcium sensitivity of the myofilament.
There were no alterations in the abundance and phosphorylation
status of the calcium cycling proteins. Figure 2 summarizes
the differences between the two isoforms.
In summary, PKC activation in cardiac hypertrophy and
heart failure has been demonstrated at the level of
neonatal cardiomyocytes in conventional models of heart
failure. Genetically-engineered mice with overexpression
in an isoform-specific manner of PKC-beta can produce
cardiac hypertrophy and failure, whereas PKC-epsilon
produces a normal form of cardiac hypertrophy.
An elegant study that interfered with Gq
function showed that LV hypertrophy in response to banding
was substantially prevented. Isoform-specific abnormalities
of PKC occur in human heart failure. PKC is activated
by diverse pathologic stimuli, including stress, ischemia,
hypoxia and reactive oxygen species, all of which play
a role in the development of heart failure. Figure 3
illustrates the overall conceptual framework. Angiotensin,
endothelin and phenylephrine signal through the PKC
pathway. Oxidative stress and stretch may directly activate
PKC. These stimuli, acting in concert, can produce cardiomyocyte
hypertrophy, pathologic hypertrophy and contractile
depression and heart failure. |
PAGE
TOP
|
Molecular
Therapeutic Targets |
| Potential therapeutic targets
in heart failure include GPCR inhibition, which acts
in part via a decrease in PKC activation. This is being
done with ACE inhibition and AT-1 receptor blockade.
Gq
inhibition has been done in the genetically-engineered
mouse, but is not likely to be used clinically because
of the beneficial effects of some of the PKC isoforms.
Isoform-specific PKC inhibition, for example with PKC-beta,
is an attractive therapeutic target. Activation of the
extrinsic off switches in a generic way might be possible.
RACK inhibition of the pathologic PKC isoform activation
is a particularly attractive target. RACKs (receptors
for activated C kinases) are molecules that act as intracellular
receptors or docking sites for PKC and may define or
at least contribute importantly to PKC isoform specificity.
Isoform-specific PKC activation, by contrast, using
a similar strategy, may be possible for PKC-epsilon.
Among the subcellular deleterious targets for enhanced
oxidative stress, which is known to occur in heart failure,
is activation of PKC. |
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2001
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|

