 |
|
|
|
| Cardiovascular Physiome: Linking
Genes and Molecules to Function |
|
Seiryo Sugiura
University of Tokyo, Tokyo,
Japan
Susumu Minamisawa
The Heart Institute of Japan,
Tokyo, Japan
Tomie Kawada
Niigata University Medical
Hospital, Niigata, Japan |
|
|
|
|
|
|
|
|
Molecular
Physiology of Myosin |
|
Using an in vitro motility assay system in an animal
model, University of Tokyo investigators studied the
role of myosin light chain in the motor function of
cardiac myosin. The experiment involved the monitoring
of the force induced by a single actin filament resulting
from its interaction with myosin.
Seiryo Sugiura and colleagues initially compared
the cardiac myosin isoforms V1 and V3, which are known
to have distinct ATPase activity based on the heavy
chain structure. They found that each force-producing
event is shorter for the V1 isoform, which displays
high ATPase activity. Next, using an in vitro expression
system, the investigators studied the mutant myosins
implicated in familial hypertrophic cardiomyopathy
and found that the gene locus, which is close to the
light chain interface, impacts heavily on motor function.
This locus is relatively far from other functionally
important domains such as the actin binding site.
Inspired by these results, Dr. Sugiura then studied
the effect of atrial and ventricular light chains
on the motor function of myosin in the rat model.
The ATPase activity of these two isoforms as measured
in solution was similar, but atrial myosin translocated
the actin filaments faster and generated less force
than ventricular myosin. The atrial myosin propelled
the actin filament about 20% faster, Dr. Sugiura reported.
The experiment also showed that the longer the actin
filament length, the more the myosin molecule participated
in the generation of more force. According to the
slope of this relationship, the ventricular isoform
produced more force (higher time-averaged crossbridge
force) than the atrial myosin. The question then became
whether the greater force generated by ventricular
myosin was due to a higher amplitude of each mechanical
event or to its longer attachment time. Further experiments
involving displacement under low and high load conditions
showed that the distribution of force amplitude was
similar between the two isoforms, however, the duration
of each mechanical event was slightly longer for the
ventricular myosin, as was the attachment time.
The mechanisms of this phenomenon remain unknown,
however, it is possible that an essential light chain
can form a link between thin and thick filaments to
modulate the actomyosin interaction. The catalytic
activity of myosin could be mainly determined by the
heavy chain structure, but the myosin light chain
also plays an important role as the modulator of kinetics,
the investigators believe.
|
PAGE
TOP
|
Calcium Cycling
Defects and Cardiomyopathy |
|
More than 18 genes have been associated with the
occurrence of cardiomyopathy. The link between the
genes and cardiomyopathic phenotypes is thought to
be a calcium cycling defect. A Japanese/US multicenter
study reported at this meeting explored possible mechanisms
for this association.
The sarcoplasmic reticulum (SR) is integral to cardiac
excitation-contraction coupling. There is re-uptake
of calcium into the SR by SR calcium ATPase (SERCA2a),
and decreased SERCA2a activity is a common feature
of cardiomyopathy. To test the therapeutic value of
enhancing calcium uptake, Susumu Minamisawa and colleagues
generated several mutant phospholamban genes that
disrupt the interaction between SERCA2a and phospholamban
(phosphalamban is an endogenous inhibitor of SERCA2a).
In genetically altered mice, phospholamban gene ablation
prevented the development of cardiomyopathy functionally
as well as morphologically. These results indicated
that calcium cycling defects are critical in the progression
of cardiomyopathy and that genetic defects can be
linked to cardiomyopathic phenotypes, said Susumu
Minamisawa, of the Heart Institute of Japan, Tokyo.
Looking for additional candidate genes, the investigators
used direct sequencing analysis to explore mutations
and single nucleotide polymorphisms of the SERCA2a
and phospholamban genes in 99 cardiomyopathic patients.
They found a single nucleotide transition, A to G
at -77, of the phospholamban promoter region and two
mutations of the SERCA2a gene in patients with hypertrophic
cardiomyopathy. One mutation of the SERCA2a gene was
a missense mutation in the cytoplasmic region, and
the other was a single nucleotide transition, C to
A, at the 5’ untranslated region. These mutations
were not found in 131 control subjects, suggesting
that SERCA2a and phospholamban are possible causal
genes for hypertrophic cardiomyopathy.
The investigators then generated a pseudophosphorylated
phospholamban mutant by replacing the serine 16 phosphorylation
site with the basic amino acid glutamine, thereby
introducing a negative charge at position 16 (S16E
phospholamban mutant). The concept was verified when
a transgenic serine 16/glutamate
mutant mouse model displayed hypercontractility and
relaxation when compared with control animals. Furthermore,
in single cardiac cells the expression of the S16E
phospholamban led to the improvement of contractility
and relaxation in DCM-model mouse ventricular cells
in the absence of catecholamines.
The investigators then tested whether the S16E phospholamban
mutant improves in vivo cardiac function of the failing
heart in a cardiomyopathic hamster model. The study
used a new gene transfer technique that involved general
hypothermia and intracoronary infusion of a recombinant
adeno-associated viral vector. The percent fractional
shortening of the left ventricle was better in the
S16E phospholamban gene-transfected hearts, even at
7 months after the gene transfer.
|
PAGE
TOP
|
Treatment
of Heart Failure by Disruption of Dystrophin-Related
Proteins |
|
Congestive heart failure is commonly caused by the
disruption of the genome or physiome of dystrophin
or sarcoglycan, a component in dystrophin-related
proteins, according to a multicenter study presented
by Tomie Kawada, of Niigata University Medical Hospital.
Dr. Kawada and colleagues reached this conclusion
by studying three congestive heart failure models:
(1) TO-2 strain hamsters with dilated cardiomyopathy
undergoing gene therapy; (2) a rat model of acute
congestive heart failure due to high-dose administration
of isoproterenol; (3) a rat model of chronic heart
failure secondary to previous myocardial infarction.
TO-2 strain hamsters with dilated cardiomyopathy
demonstrate clinical symptoms identical to humans,
in which the 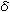 sarcoglycan gene is deleted. Sarcoglycan is a component
of dystrophin-related proteins, and its deletion and
corresponding protein dysfunction commonly induce
muscle degeneration, as is the case in muscular dystrophy.
The study used a recombinant adeno-associated virus
(rAAV) vector that included normal sarcoglycan or
Lac Z genes, driven by a cytomegalovirus promoter.
The rAAV vectors were intramurally administered to
the apex and left ventricular free wall of the 5-week-old
hamsters during open-chest surgery. At 10 or 20 weeks,
the animals underwent hemodynamic measurements, histological
examination, immunostaining, and Northern and Western
blottings and echocardiography at 30 weeks. The animals
were followed for up to 40 weeks.
sarcoglycan gene is deleted. Sarcoglycan is a component
of dystrophin-related proteins, and its deletion and
corresponding protein dysfunction commonly induce
muscle degeneration, as is the case in muscular dystrophy.
The study used a recombinant adeno-associated virus
(rAAV) vector that included normal sarcoglycan or
Lac Z genes, driven by a cytomegalovirus promoter.
The rAAV vectors were intramurally administered to
the apex and left ventricular free wall of the 5-week-old
hamsters during open-chest surgery. At 10 or 20 weeks,
the animals underwent hemodynamic measurements, histological
examination, immunostaining, and Northern and Western
blottings and echocardiography at 30 weeks. The animals
were followed for up to 40 weeks.
In vivo transfection of the rAAV vector with the
normal
sarcoglycan gene to the TO-2 hearts induced efficient
expressions of both transgene and transcript in the
myocardium. These results differed from Northern and
Western blottings seen in control hamsters and in
animals transfected only with the reporter gene. Hemodynamic
indices by cardiac catheterization showed findings
indicative of dilated cardiomyopathy in the TO-2 hamsters,
but the delta-sarcoglycan transduction improved the
parameters as compared to the group of animals transfected
with the reporter gene only. The
sarcoglycan transduction reduced the enlarged systolic
dimension, and improved both the fractional shortening
and ejection fraction accordingly.
Double fluorescence microscopy identified the myocardial
cells presenting delta-sarcoglycan or taking up exogenously
applied Evans blue dye. Cardiac muscle transduced
by the
sarcoglycan gene revealed the expression of the transgene,
but dye was not detected in this sample, indicating
the preservation of the sarcolemmal integrity. The
sarcoglycan gene treatment of the TO-2 strain hamsters
protected the cardiomyocytes from sarcolemmal leakage
in situ.
The novel gene treatment also improved survival by
Kaplan Meier analysis, as five animals in the reporter
gene group died by 35 weeks while all animals in the
delta-sarcoglycan gene replacement group survived
and remained active. The authors concluded that the
gene therapy prolonged survival because the responsible
gene causing the dilated cardiomyopathy was supplemented
in vivo. These results suggest that the genome mutation
modifies the proteome expression and finally alters
the corresponding physiome, and that the deterioration
of the physiome could be halted by the transduction
of a new genome.
In additional studies, isoproterenol caused disruption
of dystrophin but not delta-sarcoglycan. Dystrophin
was translocated to myoplasm and fragmented, while
delta-sarcoglycan was preserved in sarcolemma and
was not hydrolyzed. Dystrophin, alpha-sarcoglycan
and gamma-sarcoglycan, but not delta-sarcoglycan,
were reduced in survived myocardium after coronary
ligation. ACE inhibitors ameliorated the decrease
of dystrophin and alpha-sarcoglycan but not gamma-sarcoglycan
in vivo. Isolated m-calpain selectively degraded dystrophin,
alpha-sarcoglycan, and gamma-sarcoglycan but not delta-sarcoglycan.
These findings all suggest that congestive heart
failure is commonly caused by disruption of the genome
or physiome of dystrophin or sarcoglycan, irrespective
of hereditary or acquired origins. Somatic gene therapy
combined with pharmacotherapy is a promising approach
to preventing advanced disease.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2002
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|