|
Data from a study in patients with isolated aortic
valve stenosis conduced by Schaper and colleagues
showed that correlations of structure to function
confirm the hypothesis that transition to heart failure
occurs by fibrosis and myocyte degeneration, partially
compensated by hypertrophy involving DNA synthesis
and transcription. Cell loss, mainly by autophagy
and oncosis, contributes significantly to the progression
of left ventricular function. Because of the
limited postoperative functional recovery in later
stages, early correction of the aortic valvular defect
is recommended. This lecture reviewed their findings.
|
|
The Structure
of the Extracellular Matrix |
|
The extracellular matrix comprises a number of different
proteins. The major proteins include fibronectin,
the major matrical microfilament network all over
the extracellular matrix. Laminin and collagen IV
are the major components of the basal membrane. Collagen
I is microfibrils that stabilize the extracellular
matrix. Collagen III is a very fine filament network
surrounding the microfibrils of Collagen I. Collagen
VI sits atop this complex and is a more global component.
These proteins together with cellular structures,
such as the microvessel fibroblasts, form fibrotic
tissue. Even normal myocardium has about 10% fibrotic
tissue.
Their data were obtained in confocal microscopy
by using monoclonal or polyclonal antibodies.
In normal human myocardium, fine septa separate the
myocytes. The blast vessels are surrounded by fibronectin
because it is present in the basement membrane. In
the diseased myocardium, more fibronectin is present
and the myocytes are more separated from each other.
Replacement fibrosis is present where myocytes have
disappeared.
Collagen I is the fibular structure in the extracellular
matrix. In normal myocardium there is a small amount
of this material between the myocytes. In failing
hearts, a large amount of collagen is present. A high
amount of lipofuscin may be present in the failing
heart.
The so-called turtle projection on confocal microscopy,
obtained with special image processing, clearly shows
that collagen I is increased, in varying amounts in
the failing myocardium.
The components of the extracellular matrix are connected
with the intracellular milieu. This occurs via
three different systems: the dystrophin complex, the
spectrin complex, and the lateral costameric junction.
The extracellular matrix proteins, such as laminin
or collagen, the basal membrane, and the laminin of
the basal membrane bind to the distal glycans, and
then bind to the dystroglycans present in the sarcolemma
and represent receptors that provide connection between
the outer component and the inner part of the dystrophin
complex, which is dystrophin itself. The dystrophin
then is connected with the actin cytoskeleton and
thereby connects the extracellular matrix to the components
of the intracellular matrix, up to the nucleus, which
is very important for signal transduction.
In the spectrin complex, via the anchoring receptors,
which are ion channels in the sarcolemma connected
with the spectrin and the actin cytoskeleton. The
lateral costameric junction is a very important complex,
involving the integrins as the receptors in the sarcolemma.
The extracellular matrix proteins bind to the integrins,
which have two different subunits. These bind then
to talin, vinculin, alpha-actinin, and actin in the
intracellular milieu. Alpha-1 integrin and Beta-1
integrin are greatly increased in the failing heart.
Dystrophin localizes at the inner part of the sarcolemma.
In the healthy human heart there is nearly 400 myocytes
per square millimeter, but only half that number in
the failing myocardium. The cells in the failing heart
contain much more dystrophin than in the healthy heart,
and there is greater separation between the cells
by fibrotic material in the failing heart compared
to the healthy heart. The presence of the fibrotic
material is the first indication on the structural
level in failing myocardium that something is happening.
Some of the functions exerted by the extracellular
matrix include acting as 1) supportive scaffolding
for myocytes, myofibrils, and vasculature, 2) tethering
and proper alignment, 3) transduction of mechanical
forces, mechanical signaling, 4) provide tensile strength
and is a major determinant of compliance, 5) movement
of fluids, 6) storage of growth factors, cytokines,
latent proteases, and 7) cellular migration and adhesion.
|
PAGE
TOP
|
Study in
Aortic Valve Stenosis |
|
Schaper and colleagues examined the process of heart
failure development in patients with isolated aortic
valve stenosis. The patient population was subdivided
into 3 groups based on ejection fraction (EF): Control
group with a normal EF, Group I with a slightly elevated
left ventricular (LV) end diastolic pressure (EDP),
and an elevated pressure gradient over the aortic
wall; Group II with a significantly downregulated
EF (increased LVEDP, elevated pressure gradient);
Group III with significantly reduced EF, elevated
LVEDP, and somewhat reduced pressure gradient over
the aortic wall. The baseline characteristics are
shown in Table 1. Changes in the extracellular matrix
and in the myocyte were studied in tissue obtained
during valve replacement.
| Table
1 |
Control |
Group
I |
Group
II |
Group
III |
| Age
(years) |
68 |
69 |
69 |
72 |
| Number |
6 |
12 |
12 |
8 |
| EF
(%) |
61 |
60 |
43 |
24 |
| LVEDP
(mmHg) |
11.6 |
15.7 |
18.0 |
27.3 |
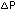 (mmHg)
(mmHg) |
11 |
63 |
55 |
42 |
Baseline
characteristics of study patients with aortic valve
stenosis. EF: Ejection Fraction; LVEDP: Left ventricular
end diastolic pressure; :
mean ventricular-aortic pressure gradient.
Changes in Extracellular Matrix
Fibrosis is significantly increased as one of the
primary events in the development of hypertrophy.
In Group I, fibrosis was significantly upregulated,
with an increase from 10% to 30% (p<0.05). Thus,
one-third of the myocardium now consisted of fibrotic
material. Remarkably, this occurred while the EF was
still normal. The patients usually do not have surgery
at this stage. In Group II, about 27% of the myocardium
was fibrotic and about 37% in Group III.
Concomitantly with the increase in fibrosis, the
number of capillaries decreased in this study. CD31,
an endothelial marker, was used to label the capillaries.
In the normal human myocardium, there were nearly
2000 capillaries per square millimeter, and this decreased
linearly with the increase in fibrosis.
Angiotensin converting enzyme (ACE) localization
in myocardium was assessed, to identify cytokines
that may be responsible for the development of fibrosis.
Most of the capillaries were labeled for ACE. There
was a sharp increase from the control group to the
study groups in the amount of ACE-positive capillaries,
affecting about 28% of all vessels.
TGF-beta was present in fibroblasts and macrophages,
and also in a stored form in the extracellular matrix
without being bound to any cellular structure. The
fluorescence intensity of TGF-beta progressively increased,
from about 25% in the control group to about 60% in
Group III. So, TGF-beta, which is required for the
development of fibrosis and for the action of ACE,
is increased during hypertrophy.
Pan-leukocytes, lymphocytes, and macrophages are
increased in the hypertrophied myocardium, suggesting
a chronic, slow inflammatory process in the myocardium.
A causal relation between the degree of fibrosis
and the degree of EF was suggested by their positive
correlation; the more fibrosis, the lower the EF.
Also, a causal relation was found between fibrosis
and LVEDP; the more fibrosis, the higher the LVEDP.
Myocyte Changes
The myocytes are comprised of different proteins,
summarized in five different protein families: contractile,
cytoskeletal, membrane-associated sarcomeric skeletal,
and intercalated disc.
Degenerative changes in a number of the myocytes
in these protein families were evaluated in the failing
myocardium by electron microscopy and confocal microscopy
in the present study. Myocyte degeneration is defined
Schaper et al as the involvement of all cellular organelles
in a chronic and most probably slow process of degradation
that finally results in cellular atrophy, cell death,
and finally in replacement fibrosis.
In this study, the degree of myocyte degeneration
was 0.5% in the control group, and 5% in Group 1,
11 % in Group II, and 16% in Group III. Importantly,
these investigators found that fibrosis occurs earlier
than damage to myocytes. Fibrosis is the primary event
in the development from hypertrophy to failure. These
are positively correlated; the greater the degree
of fibrosis, the greater the degree of myocyte degeneration.
Hence, myocyte degeneration and fibrosis probably
have an additive negative effect on the structural
integrity of the myocardium.
Myocyte degeneration also correlates with reduced
EF, with the greater the degeneration, the lower the
EF. Both myoctye degeneration and fibrosis seem
to play a very important role in causing heart failure.
Replacement fibrosis is present in the entire failing
myocardium, indicating that many cells, of different
types, must be dying. However, this group did not
find a very high number of apoptotic cells. Therefore,
they looked for other types of cells occurring in
failing human hearts.
|
PAGE
TOP
|
Types of
Cell Death in Heart Failure |
|
Multiple mechanisms for cell death exist in the failing
human heart. Three types of cell death have been identified
by Schaper and colleagues.
Apoptotic cell death is found in the human heart,
but not frequently. These TUNEL-positive cells, contain
some lipofuscin granules. The typical symptoms of
apoptosis were found in the human heart, such as condensation
of the comaten, preservation of the mitochondrial
structure, and preservation of cellular membrane.
Oncotic cell death is the second type found in the
failing human heart. This is accidental ischemic cell
death, in contrast to the programmed cell death in
apoptosis. Previously called necrotic cell death,
this and other research groups changed the name to
oncotic cell death, to identify the killing system,
not the necrosis that occurs after the cell dies.
A leaky membrane is the main characteristic of oncotic
cell death, and is the leading functional symptom.
Complement 9 staining identifies the leaky membrane
and allows the number of oncotic cells in tissue to
be counted. Also seen are lipofuscin granules and
myocytes that are still intact. Electron microsopy
reveals a swollen nucleus, clustering of the comaten,
and defective mitochondria.
Autophagic cell death is the third type of cell
death. Schaper and colleagues are the first to describe
this type of cell death in the failing human myocardium.
It has previously been found in lymphocytes or granulocytes
and cells in the thymus. Autophagic cell death involves
lysosomal protein degradation via ubiquitin-dependent
and independent pathways. It is caspase-independent
but ATP-dependent cell death. Bursch previously described
autophagic cell death as a catabolic process involving
extensive autophagic degradation of cellular organelles
and autophagic vacuoles. Autophagic cell death is
a very slow process involving the ubiquitinization
of many different proteins. Immunogold labeling of
autophagic cells revealed a high amount of ubiquitin.
|
PAGE
TOP
|
Research
Findings in Autophagic Cell Death |
|
Ubiquitin, under normal conditions, is responsible
for the degradation of proteins by making a conjugate,
a sort of complex between proteins and ubiquitin,
and then bringing this complex into the so-called
proteosome. Leading investigators of proteosomal protein
degradation state that 90% of all proteins under normal
circumstances are being degraded in the proteosome.
This requires an entire cascade to occur. First, ubiquitin
must be bound to an activating enzyme, E1, and then
to the conjugating enzyme E2. The protein is ubiquitinated
only after formation of this complex, with the help
of the ligating enzyme E3. The ubiquitin protein complex
is transported to the proteosome. Before it can enter
the proteosome, cleavage of the ubiquitin in certain
places must occur. The ubiquitin then recycles and
begins the whole process again, whereas the protein
enters the proteosome, a cylindrical structure, where
it is degraded to peptides and finally to amino acids.
This is the most important mode of protein degradation
in normal and in diseased cells.
The cascade of these enzymes must be intact and
the activity of the de-ubiquitinating enzymes must
be high for this process to occur. Otherwise, ubiquitin
protein complexes cannot enter the proteosome, in
which case it will be stored. This is likely
what occurs in the failing myocardium.
A significant increase in the ubiquitin conjugating
enzyme was found in dilated cardiomyopathy compared
to control, explaining the reason many ubiquitin protein
complexes are found in the failing heart. Ligase was
unchanged. Importantly, the deubiquitinating
enzyme isopeptidase T and the ubiquitin fusion degradation
protein one (UFD-1) were decreased in failing myocardium.
This means there is no more cleavage of the protein
ubiquitin complexes. This is second reason for the
accumulation of these complexes in the cell.
This group found that the proteosome still contains
a normal amount of protein and that the proteosomal
activity is variable among patients. But the mean
values of proteosome in the failing heart and in the
controls are similar. So, proteosomes are not involved
in autophagic cell death. But, the decrease in the
deubiquitinating enzyme can explain autophagic cell
death. Further, potepsin, another important proteolytic
enzyme, also was reduced.
|
PAGE
TOP
|
Estimating
Rates of Myocyte Cell Death in Heart Failure |
|
The time course between the initiation of a certain
type of cell death and the duration of the death cascade
must be known to estimate the rate of myocyte death,
plus when the removal of the myocyte has been completed.
For oncosis, data from animal experiments may be
transferred to the human myocardium. When assuming
that the duration of the death cascade was 45 to 60
minutes and that these cells are usually removed in
about 48 hours. in failing myocardium, 0.06% of all
cells were positive for oncosis. This translates to
11% of myocytes dying annually from oncotic cell death.
Apoptotic cell death seems not to be an important
mechanism of death in heart failure. This group estimated
that only 1.1% of myocytes die annually from apoptotic
cell death, based on experiments devised by this group.
They found that the duration of the death cascade
in apoptotic cell death is about 14 hours, after it
is initiated by H2O2 in myocyte
cultures.
Autophagic cell death has not been studied in myocardial
tissue. Thus, the annual myocyte death by autophagic
cell death is unknown. Autophagic cell death is a
very slow process, perhaps taking days or months.
But, because of the large number of degenerating myocytes
that are seen, this group believes that the process
of autophagic cell death is a very important one.
In the progression from compensated hypertrophy
to heart failure, myocyte degeneration contributes
significantly to functional deterioration. The final
stage of degeneration is cell death. Fibrosis and
degeneration have additive effects.
|
PAGE
TOP
|
|
Morphometric data from their present study showed
that the cross-sectional area of the cells increase
as failure progresses, indicating hypertrophy. The
size of the nuclei increase, but not to the degree
required to make the ratio between cell volume and
nuclear volume similar to that in the control group.
The presence of the splicing factor Sc-35 is marker
for ongoing transcription, and can thus indicate whether
or not cells are still viable. A significant
increase in the content of Sc-35 and of DNA was found
in the hypertrophied heart in the present study. This
increase was most significant in Group III. However,
this is not counterbalanced by an increase in the
concentration of DNA and of SC-35, indicating insufficient
SC-35 to ensure transcription.
|
PAGE
TOP
|
|
Fibrosis is a primary event in the development of
cardiac hypertrophy. Myocyte degeneration occurs later,
when the EF is already reduced. Low-grade inflammation
present in both hypertrophied and failing hearts.
Upregulation of ACE is an early event, whereas TGF-beta
is late. Cell death by 3 different mechanisms is evident.
The DNA content is increased in the failing heart,
but not the DNA concentration. The Sc-35 content is
increased in the failing heart, but not the concentration.
Sc-35 elevation indicates cellular viability.
These investigators propose this scheme for the
progression from pressure overload to heart failure.
Pressure overload is the primary event and causes
reactive fibrosis, in which TGF-beta and ACE are involved.
Capillary density decreases, which is ensued by myocyte
degeneration, hypertrophy, and cellular sequestration.
Low-grade inflammation and myocyte hypertrophy further
enhances this process. There are some counteracting
mechanisms in this scheme. The nuclei try to adapt
to the higher requirements of the hypertrophied cells,
but when this adaptive process becomes exhausted the
consequence is myocyte loss and then replacement fibrosis.
This creates a vicious circle, finally leading to
heart failure.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2003
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|