|
|
|
|
| Molecular Mechanisms and Therapeutic Strategy for Cardiac Remodeling |
|
|
HB-EGF is a Potential Growth Factor for Cardiomyocyte Metabolism. The Possibility for a Novel Therapeutic Target for Heart Failure.
Seiji Takashima
Osaka University Graduate School of Medicine, Osaka, Japan
Kruppel-Like Zinc-Finger Transcription Factor KLF5/BTEB2 is a Target for Angiotensin II Signaling and an Essential Regulator of Cardiovascular Remodeling
Takayuki Shindo
University of Tokyo, Tokyo, Japan
Critical Roles of Fas/Fas Ligand Interaction and Beneficial Effect of Soluble Fas Gene Therapy for Post-Infarct Ventricular Remodeling and Dysfunction
Genzou Takemura
Gifu University School of Medicine, Gifu, Japan
Role
of Proinflammatory Cytokines in Cardiac Remodeling
Toru Kubota
Kyushu University Graduate School of Medical Sciences , Fukuoka, Japan
Mammalian Target of Rapamycin (mTOR): A New Molecular Target for Cardiac Hypertrophy
Tetsuo Shioi
Kitasato University School of Medicine, Sagamihara, Japan
Therapeutic Myo-Angiogenesis for Ischemia-Induced Myocardial Remodeling by Transplantation of Autologous Bone Marrow Mononuclear Cells
Hiroaki Matsubara
Kansai Medical University, Moriguchi, Japan
|
|
|
|
|
HB-EGF is a Potential Growth Factor for Cardiomyocyte Metabolism. The Possibility for a Novel Therapeutic Target for Heart Failure.
Seiji Takashima
Osaka University Graduate School of Medicine, Osaka, Japan
|
|
Heparin binding EGF-like growth factor (HB-EGF),
one of the EGF family growth factors, was first discovered
as a fibroblast growth factor from macrophage conditioned
media. It is secreted as a membrane-anchored form,
and truncated by processing enzyme to exert its activity
through EGF receptor binding. This HB-EGF processing
mechanism is important for cardiac hypertrophic signaling
by GPCR agonist. These investigators developed an
HB-EGF transgenic mouse to clarify the role of HB-EGF
processing.
EGF-receptor transactivation is mediated in two ways:
1) intracellular signaling molecules, such as SERC,
and 2) via an alternative pathway, in which a membrane-bound
EGF-family ligand metalloproteinase inhibitor is activated,
binds soluble EGF to the EGF receptor from the outside
and transactivates the EGF receptor.
The alternative transactivation also occurred in cultured
rat cardiomyocytes and is involved in cardiac hypertrophic
signaling mediated by catecholamine, angiotensin,
and endothelin. These neurohormonal factors bind to
the receptor and transactivate metalloproteinase,
and thus transactivate the EGF receptor. The metalloproteinase
inhibitor KB-R7885 inhibits this transactivation and
the cardiac hypertrophy induced by the neurohormonal
factors. Sharp19, the HB-EGF neutralizing antibody,
inhibited the transactivation by the neurohormonal
factors and also protein synthesis. The metalloproteinase
inhibitor KB-R7885 is also active in vivo, and has
been shown to inhibit cardiac hypertrophy induced
by continuous infusion of angiotensin II and endothelin.
Thus, these investigators concluded that neuorhormonal
factors activate metalloproteinase and shed the membrane-anchored
HB-EGF, and then soluble HB-EGF binds to the EGF receptor
and induces cardiac hypertrophy.
Their subsequent work in wild type and HB-EGF pro/pro
mice showed that 1) blocking HB-EGF shedding from
cell membranes causes severe heart failure and early
death by cardiomyocyte cell death in the HB-EGF pro/pro
mice, 2) in HB-EGF del/del mice the HB-EGF null mouse
shows a similar phenotype with the HB-EGF pro/pro,
and 3) both phenotypes were similar to human dilated
cardiomyopathy (DCM). Interestingly, none of the other
ligands in the EGF family show this cardiac phenotype.
Although HB-EGF function was abrogated in the entire
body in HB-EGF pro/pro and HB-EGF del/del mice, only
the heart was affected. The investigators concluded
that HB-EGF is an essential growth factor in cardiac
myocyte metabolism.
|
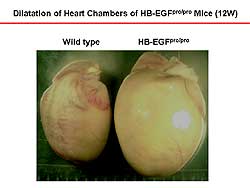 |
| Figure
2. The heart of the HB-EGF transgenic mice is
enlarged at 12 weeks compared to the wild-type
mice. |
| Click
to enlarge |
|
To examine the in vivo role of HB-EGF the investigators
made a targeting mouse in which the HB-EGF is not
truncated from the cell membrane. In the targeting
construct, the exon 1 through 3 region was replaced
by HB-EGF full-length CDNA, which has a 2-point mutation
that does not allow the HB-EGF to become truncated
from the cell membrane and remains as HB-EGF in transmembrane
form (Figure
1). The homozygote mouse expressing uncleavable
mutant HB-EGF is called HB-EGF pro/pro mouse. The
HB-EGF pro/pro mouse was healthy at birth. The promoter
region of the HB-EGF pro/pro mouse was intact and
it expressed the same amount of mutant HB-EGF at the
same organ as did the wild-type mouse. Wild-type HB-EGF
was null in the HB-EGF pro/pro mouse. The HB-EGF pro/pro
mouse died early compared to the wild-type mouse,
with one-half dead at 120 days.
At 12-weeks, the body size and weight of the wild-type
and HB-EGF pro/pro mouse were nearly the same. Although
the HB-EGF mutant was expressed in the entire body,
the abnormal phenotype was observed only in the heart.
The heart size was enlarged (Figure
2) and the lung weight increased and pulmonary
infusion was detected in the HB-EGF pro/pro mice,
indicating death was from heart failure.
|
|
Echocardiographic analysis indicated that the heart
of the HB-EGF pro/pro mouse was enlarged and fractional
shortening was reduced remarkably. Blood pressure
was slightly lower than in wild-type mice, but the
heart rate was comparable. The extracted heart showed
remarkable enlargement in size, and the coronary vessel
was intact in HB-EGF pro/pro mice. Cross-section of
the LV chamber showed enlargement of chamber size.
Histologically, HB-EGF pro/pro showed massive degradation
of cardiomyocytes and repression of fibrosis. The
coronary vessel was nearly intact. High-magnification
HE staining showed massive myocyte degradation, which
was repressed by fibrosis. No leukocyte infiltration
was seen, indicating an autoimmune or infectious mechanism
was not involved in this phenotype.
Histological changes at 6 weeks in the HB-EGF pro/pro
mouse were hypertrophic cardiomyocytes and detection
of vacuole formation. Cross-sectional examination
showed that the vacuole was pressed on the intercalated
disk, and further examination showed it to be a dissection
of the intercalated disk. Similar vacuole formation
and intercalated disk dissection was observed in human
DCM biopsy samples.
In a HB-EGF null mouse (HB-EGF del/del) mouse, in
which HB-EGF CDNA was knocked out the whole body,
HB-EGF expression was abrogated in the entire body.
The null mouse showed a similar phenotype to the HB-EGF
pro/pro mouse that had a dilated heart chamber and
reduced contractility.
Thus, the investigators hypothesize that HB-EGF is
partly mediating the cardiac hypertrophy signals through
EGF-receptor phosphorylation. However, it is completely
lost towards the cardiac cell death, indicating that
HB-EGF is essential for cardiomyocyte metabolism.
So, for heart failure treatment, drugs that block
this pathway are being used, with their beneficial
effect probably mediated by reducing contractility,
heart rate, and blood pressure. However, adding HB-EGF
or another signaling molecule may be beneficial for
the survival of the cardiomyocyte. The combination
of a blocking agent, such as beta blocker or angiotensin
II blocker, and EGF or another growth factor, may
be a better therapeutic way to treat heart failure.
|
PAGE
TOP
|
Kruppel-Like Zinc-Finger Transcription Factor KLF5/BTEB2 is a Target for Angiotensin II Signaling and an Essential Regulator of Cardiovascular Remodeling
Takayuki Shindo
University of Tokyo, Tokyo, Japan
|
|
The transcription regulation and molecular links
between stress and cardiovascular (CV) remodeling
remain to be clarified. The novel transcription factor
KLF5/BTEB2 was discussed in this lecture.
Phenotypic modulation of smooth muscle cells from
the contractile to synthetic type is important for
the pathogenesis of atherosclerosis. In this process,
SMemb gene expression is induced. This group originally
identified KLF5 as a transcription factor that binds
to the promoter region of the SMemb gene and upregulates
its transcription. KLF5 is a member of a Kruppel-like
transcription factor, and possesses a transcription
activating domain and DNA binding Zn-finger motif.
KLF5 is highly expressed in the cardiac smooth muscle
cells or during development in the vascuole, but is
downregulated in adults. In contrast, KLF5 induces
activated smooth muscle cells in the neointima. In
the rabbit balloon injury model, high expression of
KLF5 is detected in the neointima. KLF5 transactivates
various genes in addition to SMemb in vitro, for example,
Egr-1, PAI-1, iNOS, and VCAM. Thus, KLF5 might be
involved in cardiovascular (CV) lesions through the
activation of these factors.
To better understand the in vivo function of KLF5,
Shindo and colleagues generated a KLF5 knockout mouse.
KLF5 is essential for fetal development; homozygotes
died in utero prior to 8.5 dpc. Heterozygotes are
apparently normal and fertile. KLF5 knockout mice
seem to have thinner vascular walls, with thinning
of the medial and advential layers. In spite of these
changes, no major abnormalities were found in the
tension development of the aortic ring by angiotensin
II (Ang II) or endothelin 1 (ET-1) or in systolic
blood pressure.
In the cuff injury model of the femoral artery, after
5 weeks formation of granulation tissue and microvessels
was detected around the polyethylene cuff in wild-type
mice, but was limited in the knockout mice. The arteries
of the KLF5 knockout mice were thin-walled and dilated,
in striking contrast to the thickened medial and intimal
layers in the wild-type mice. The area of neo-intima
formation was less than 2000 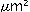 in the knockout mice, compared to 12000
in the wild-type mice. KLF5 is important for angiogenesis
as well as neointima formation.
in the knockout mice, compared to 12000
in the wild-type mice. KLF5 is important for angiogenesis
as well as neointima formation.
|
|
|
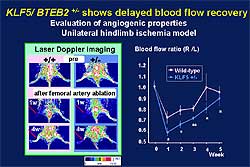
Angiogenesis is reduced in the KLF5 knockout mice,
as shown by the delayed vessel recovery in the unilateral
hindlimb ischemia model (Figure
1). In the tumor transplantation model using Sarcoma
180 (S180), tumor growth at 2 weeks, blood flow, and
capillary formation were reduced in the knockout mice.
In the Ang II infusion model, no differences were
detected after 14 days in the systolic blood pressure
or angiotensin II receptor expression levels. But,
cardiac hypertrophy, perivascular fibrosis, and interstitial
fibrosis were reduced in the knockout mice. KLF5 is
a critical transcription factor for cardiovascular
remodeling, including cardiac hypertrophy, fibrosis,
angiogenesis, and atherosclerosis, based on these
data.
|
|
Downstream factors in the pathogenesis of
CV remodeling
PDGF-A expression is selectively reduced in KLF5 knockout
mice. KLF5 is also abundantly expressed in the intestinal
tract. Karlson and colleagues showed that KLF5 shows
abnormal structure of the digestive tract, such as
misshapen villi, reduction in the number of mesenchymal
cells, and reduction in the amount of extracellular
matrix in knockout mice. Notably, this phenotype is
very similar to those of PDGF and knockout mice.
Shindo and colleagues analyzed the relation between
KLF5 and PDGF-A. Upregulation of KLF5 was detected
2 hours after Ang II-stimulation of cardiac fibroblast
and continued over a 4-hr period, and then was decreased
because of PDGF-A regulation.
KLF5 overexpression significantly upregulated the
transcription of PDGF-A (Figure
2). In Ang II-stimulated cardiac fibroblasts,
KLF5 was shown to directly bind to the PDGF-A promoter
in an Ang II-dependent manner (Figure
3).
|
|
 |
| Figure
2. KLF5 overexpression significantly upregulated
the transcription of PDGF-A on a reporter assay
of PDGF-A. |
| Click
to enlarge |
|
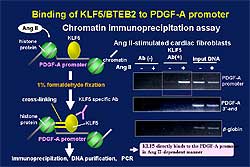 |
| Figure
3. KLF5 binds directly to the PDGF-A promoter
in an Ang II-dependent manner. |
| Click
to enlarge |
|
 |
| Figure
5. The characteristics of AM80, a possible modulator
of KLF5. |
| Click
to enlarge |
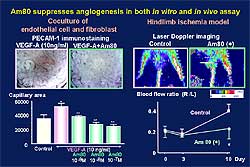 |
| Figure
6. AM80 suppressed angiogenesis in in vitro and
in vivo assays. |
| Click
to enlarge |
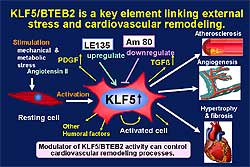 |
| Figure
7. KLF5 links external stress and cardiovascular
remodeling. |
| Click
to enlarge |
|
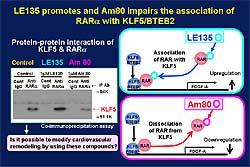
Modifying CV remodeling
The administration of LE135 to KLF5 knockout mice
in the cuff injury model increased granulation tissue
and neointimal formation, while these were decreased
with the administration of AM80 to wild-type mice.
The characteristics of AM80, a possible modulator
of KLF5, are shown in Figure
5. In a clinical trial of 66 patients with acute
promyelocytic leukemia, AM80 was associated with an
81% remission rate. AM80 suppressed angiogenesis in
in vitro and in vivo assays, as shown in Figure
6. Capillary formation enhanced by PDGF-A was
suppressed dose-dependently by AM80. In the hindlimb
ischemia model, angiogenesis was reduced in AM80-treated
mice. In a tumor transplantation model with S180,
tumor weight, tumor blood flow, and tumor capillary
formation were reduced in AM80-treated mice. AM80
also suppressed cardiac hypertrophy. In the Ang II
infusion model, AM80-treated mice had thinner ventricular
wall on M-mode echocardiography.
Based on these data, these investigators conclude
that KLF5 is a key element linking external stress
and cardiovascular remodeling (Figure
7). Drugs that target KLF5 are effective to control
CV remodeling. Modulation of KLF5 activity can control
CV remodeling including atherosclerosis, angiogenesis,
cardiac hypertrophy, and fibrosis.
|
PAGE
TOP
|
Critical Roles of Fas/Fas Ligand Interaction and Beneficial Effect of Soluble Fas Gene Therapy for Post-Infarct Ventricular Remodeling and Dysfunction
Genzou Takemura
Gifu University School of Medicine, Gifu, Japan
|
|
The preservation of granulation tissue cells of
subacute myocardial infarction (MI) through blockade
of apoptosis may result in the improvement of post-infarction
left ventricular remodeling and heart failure, hypothesized
these investigators based on the findings of their
previous work.
In a rat model of MI, a pan-caspase inhibitor (BAF)
effectively blocked apoptosis of myocardial granulation
tissue cells during the subacute stage of infarction.
In the granulation tissue of 2-week post-infarction
rats, apoptosis of endothelial cells, myofibroblasts,
and macrophages were identified on electronmicroscopy.
TUNEL positive cells were significantly reduced in
the BAF-treated rat hearts compared to control infarct
hearts. The apoptotic index of the control infarcted
heart was 1.87% while that of BAF-treated heart was
0.69%. The total number of interstitial cells was
significantly greater in the infarct area of BAF-treated
hearts, probably reflecting the reduced apoptosis
of these cells.
At 12 week, the rats treated with BAF for the first
4 weeks had a higher survival rate compared to the
control rats, had greater left ventricular (LV) wall
thickness, smaller LV end diastolic diameter, and
greater percent fraction shortening. Treatment with
the caspase inhibitor improved LV remodeling and cardiac
contractility of the post-infarct heart in the chronic
stage. BAF treatment also relieved congestion and
improved cardiac performance of the LV of the post-infarct
heart during the chronic stage, as shown by the lower
central venous pressure and LV end diastolic pressure
and greater LV dPdt.
Severe post-infarct LV remodeling was attenuated by
BAF treatment, and the body weight to heart weight
ratio was significantly decreased. BAF-treated rats
had a thicker infarct wall containing collagen fibers
and abundant cellular components and many small vessels
compared to control rats, which had thinner walls
with fibrous scar tissue containing scanty cell component.
The non-myocyte cell population was more than 2-fold
greater in the BAF-treated rats compared to control
rats. The percent area of  -smooth
muscle actin positive cells in the infarct area was
significantly greater in the BAF-treated rat hearts
than in the control. -smooth
muscle actin positive cells in the infarct area was
significantly greater in the BAF-treated rat hearts
than in the control.
The number of vessels obtained was about 3-fold greater
in the treated heart compared to the control. In contrast,
the macrophage, a major cell component of granulation
tissue, was not preserved at 12 weeks post-infarction.
Electromicroscopic examination revealed myofibroblasts
and many groups of mature smooth muscle cells with
contractile phenotype in the BAF-treated rats. These
smooth muscle cells may originate from the preserved
myofibroblasts.
Although the pancaspase inhibitor was found to beneficially
effect post-infarct remodeling and dysfunction, there
are several limitations of this reagent. Apoptosis
of many cell types, both physiological and pathological
apoptosis, is largely caspase-dependent. Thus, possible
adverse effects by apoptosis inhibition may occur,
even if used for a short time. This study did not
refer apoptotic machinery specific for granulation
tissue cells of MI. Hence, a more specific way to
inhibit granulation tissue cell apoptosis is desirable.
|
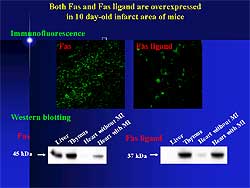 |
| Figure
1. Fas and Fas ligand are overexpressed in the
post-infarct granulation tissue cell of mice. |
| Click
to enlarge |
 |
| Figure
2. TUNEL positive cells were reduced in the soluble
Fas treated mouse hearts. |
| Click
to enlarge |
|
The Fas ligand system is important during the initial
stage of apoptosis. Both Fas and Fas ligand are overexpressed
in the post-infarct granulation tissue cell of mice
(Figure
1). The lymphoproliferative (Lpr) mouse strain
congenitally lacks Fas and Fas interaction, because
of Fas gene defects. The generalized lymphoproliferative
disease (Gld) strain contains a point mutation in
the Fas ligand gene, thus Fas ligand is non-functioning.
In an MI model of these strains, at 4 weeks post-MI,
ventricular remodeling was attenuated and cardiac
function was improved.
This group then showed that soluble Fas interferes
with Fas and Fas ligand interaction and thus is an
inhibitor of Fas-mediated apoptosis. They generated
a recombinant adenoviral vector of mouse soluble Fas
genes. On day 3 post-MI, the adenoviral vector was
injected into the hindlimb. The control mice were
injected with LacZ gene. The level of soluble Fas
in the plasma reached 31.8 mg/ml at 4 days after viral
injection. The normal range in humans of soluble Fas
is about 2 nanogram/ml. The results were strikingly
similar to the case of the treatment with pancaspase
inhibitor.
TUNEL positive cells in the granulation tissue post-infarction
were significantly reduced in the soluble Fas treated
mouse hearts compared with control (Figure
2) At the chronic stage of MI, LV end diastolic
and end systolic diameters were smaller and the percent
fractional shortening was greater in the treated hearts.
|
|
Soluble Fas attenuated post-infarct ventricular
remodeling and dysfunction in the treated mice (Figure
3). The body weight to heart rate was significantly
decreased and the infarct wall was thicker in the
treated compared to the control mice.
The Flk-1 positive vessels were preserved more abundantly
in the infarct of the soluble Fas treated hearts.
In the control infarct tissue, -smooth
muscle actin positive cells were rarely observed,
but they were preserved in the infarct area of the
soluble Fas treated hearts. However, macrophages were
equally rare in both groups. Interestingly, there
were bundles of smooth muscle cells with the contractile
phenotype in the old infarct area. The beneficial
effects of the soluble Fas treatment were observed
at 10 weeks post-MI (Figure
4).
|
|
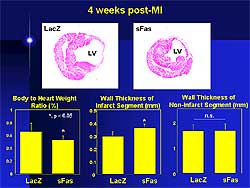 |
| Figure
3. Effects of soluble Fas and LacZ post myocardial
infarction. |
| Click
to enlarge |
|
|
 |
| Figure
5. Factors involved with the beneficial effects
of anti-apoptotic treatment of granulation tissue
cells. |
| Click
to enlarge |
|
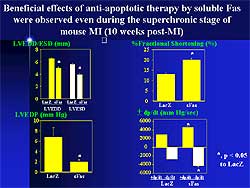
In another experiment, anti-apoptotic therapy was
begun 3 weeks post-MI. At 10 weeks, no beneficial
effect of soluble Fas therapy was found on LV remodeling
and dysfunction. This negative finding confirms the
concept that granulation tissue cell apoptosis critically
influences the post-infarct ventricular remodeling
and dysfunction.
Figure
5 illustrates the factors considered responsible
for the beneficial effects of anti-apoptotic treatment
of granulation tissue cells. Wall stress is an important
factor that accelerates ventricular remodeling and
is directly proportional to wall thickness. Thus,
a thick infarct wall as a result of anti-apoptosis
therapy can reduce ventricular wall stress. Therapy
may interfere with the vicious cycle between wall
stress and remodeling. Bundles of smooth muscle cells
with contractile phenotype running in parallel with
the surviving myocytes might help cardiac contractility.
Preservation of vessels might relieve ischemia of
the surviving tissue.
In summary, the Fas and Fas ligand interaction is
critical in the apoptosis of granulation tissue cells
after MI. Soluble Fas gene therapy begun on day 3
post-MI alleviated post-infarct LV remodeling and
heart failure at the chronic stage. The present finding
may imply a novel therapeutic strategy against chronic
progressive heart failure after a large MI, and can
be given to patients who have lost the opportunity
for coronary reperfusion therapy at the acute stage.
|
PAGE
TOP
|
Role
of Proinflammatory Cytokines in Cardiac Remodeling
Toru Kubota
Kyushu University Graduate School
of Medical Sciences , Fukuoka, Japan
|
|
Myocardial production of proinflammatory cytokines,
especially tumor necrosis factor alpha (TNF-)
is increased in patients with heart failure. To investigate
the role of proinflammatory cytokines in the myocardium,
Kubota and colleagues developed transgenic mice with
cardiac-specific overexpression of TNF-.
The transgene was driven by murine -myosin
heavy chain (-MHC)
promoter to ensure cardiac overexpression.
Previous work by this group has shown that overexpression
TNF-
causes: myocardial inflammation with extracellular
matrix remodeling, ventricular hypertrophy, with four-chamber
dilation, impaired contractility with diminished ß-adrenergic
inotropic responsiveness, reactivation of the fetal
gene program with down-regulation of calcium handling
genes, and premature death with heart failure. These
results indicate, they conclude, that myocardial production
of TNF-
may play an important role in the pathogenesis of
myocardial dysfunction and ventricular remodeling.
However, the mechanism by which TNF-
induces cardiac remodeling and dilated cardiomyopathy
is unclear. Data from this group shows that nitric
oxide (NO) exerts versatile effects on cardiovascular
(CV) function. Recent studies have shown that iNOS
is increased in the failing human heart. A small amount
of NO produced by nNOS and eNOS seems cardioprotective
by improving myocardial perfusion and inhibiting apoptosis.
In contrast, a large amount of NO produced by iNOS
may be cardiotoxic by suppressing myocardial contractility
and promoting apoptosis. These investigators hypothesize
that since TNF-
is a potent inducer of iNOS, the negative inotropic
effect of TNF-
may be mediated by the enhanced production of NO in
the myocardium.
They first investigated the effects of iNOS inhibition
on cardiac function, using the selective iNOS inhibitor
ONO-1714. Blood pressure or heart rate was not changed
with ONO-1714 in wild-type or transgenic mice. Although
ONO-1714 did not affect baseline contractility estimated
by dP/dt max, it significantly improved beta adrenergic
inotropic hyporesponsiveness in TNF-
transgenic mice, but not in wild-type mice in which
iNOS was not unregulated.
Because iNOS inhibition significantly improved beta
adrenergic inotropic responsiveness in transgenic
mice, these investigators hypothesized that knocking
out iNOS may improve cardiac function and prolong
survival of TNF-
transgenic mice. In TNF-alpha transgenic mice crossed
with iNOS knockout mice, eNOS expression was not affected
but iNOS activity was completely abolished in the
myocardium. Blood pressure or baseline contractility
was not affected in this double crossover model. However,
disruption of the iNOS gene improved ß-adrenergic
inotropic responsiveness only in TNF-
transgenic mice.
No change in interstitial infiltration was seen with
NO. Cytokine expression was not affected by iNOS knockout.
In the iNOS knockout mice, heart failure development
and survival was not changed, indicating that although
myocardial expression of iNOS plays a key role in
the attenuation of ß-adrenergic inotropic responsiveness,
NO-independent mechanisms might be more important
in the development of heart failure (Figure
1).
|
PAGE
TOP
Mammalian Target of Rapamycin (mTOR): A New Molecular Target for Cardiac Hypertrophy
Tetsuo Shioi
Kitasato University School of Medicine, Sagamihara, Japan
|
|
Cardiac hypertrophy, typically defined as increased
myocardial size, is a major risk factor for heart
disease. The role of insulin in the phosphoinositide
3-kinase (PI3K) pathway in determining myocardial
size and the mammalian target of rapamycin (mTOR)
in cardiac hypertrophy was reviewed.
The mechanism of organ size regulation has been studied
in several models. Work on the wing size of Drosophila
suggests that organ size is not regulated at the cellular
level, but at the organ level. All of the genes identified
to control organ size in Drosophila are in the PI3K
pathway. Most of the genes control organ size by regulating
both cell size and cell number in the organ, but some
regulate organ size by regulating only cell size.
|
|
|
Insulin and insulin growth factor receptors phosphorylate
insulin receptor substrates (IRS). Phosphorylated
IRS activate PI3K. Expression of activated PI3K in
Drosophila wing promotes wing growth and expression
of dominant-negative PI3K mutant results in a smaller
wing. Other developmental processes are not disturbed.
The role of insulin and insulin-like growth factor
1 (IGF 1) in physiological and pathological conditions
have been studied extensively (Figure
1).
To study the role of the insulin signaling pathway,
his group moderated the activity of the members of
the pathway by genetic methods. IGF 1 receptor gene
was overexpressed in a heart-specific manner. Heart
size was increased in IGF 1 receptor mice, with increased
wall thickness while the proportion of the size of
each chamber was preserved. Cardiomyopathic changes,
such as necrosis or fibrosis, were not observed. The
mice survived normally and cardiac function was preserved
on echocardiography.
|
|
They generated transgenic (TG) mice that expressed
contributive active PI3K or dominant-negative PI3K
in a heart-specific manner. Expression of contributive
active PI3K resulted in a larger heart, and dominant-negative
PI3K resulted in smaller heart.
Increase or decrease in heart size was associated
with a comparable increase or decrease in cardiac
myocyte cell size. All of the mice survived normally
and cardiac function was preserved on echocardiography,
even in the smaller hearts.
They also characterized cardiac specific PTEN knockout
mice. Antagonism of PI3K function resulted in Akt
activation in the heart tissue of the knockout mice.
Heart weight was increased by 35% in PTEN knockout
mice. This group has shown that IGF-1 receptor, PI3K,
PTEN, and Akt are important in heart size examination.
Hence, the insulin-PI3K pathway is an important signaling
module for organ size regulation in mammals. To identify
target genes of PI3K in intact heart tissue, they
prepared RNA from heart tissue from PI3K TG mice and
performed CDNA chip analysis. If a gene is upregulated
or downregulated in dominant-negative PI3K mice and
the gene is regulated in the opposite way in constitute
active PI3K mice it is very likely that the gene is
a direct target of PI3K. For example, cardiotropin
gene is likely to be regulated by PI3K. Cardiotropin
is an important growth factor for cardiomyocyte. So,
PI3K may regulate heart size by regulating such growth
factors. Work by Izumo and colleagues shows that a
single nucleotide polymorphism (SNP) of the IGF 1
gene appears to be associated with heart size, suggesting
that insulin signaling may be involved in cardiac
hypertrophy in humans.
This group showed in transgenic mice studies that
the insulin signaling pathway is important in the
development of heart growth. To examine the role of
the pathway in cardiac hypertrophy induced by pathological
stress, rapamycin was used as a pharmacologic probe.
Rapamycin inhibits the activity of mTOR, which regulates
downstream effectors of insulin signaling pathways.
4E-BP1 regulates the activity of the translation-initiation
complex. Liposomal S6 kinase (S6K) is another important
target of mTOR. S6K phosphorylates 40S ribosomal S6
protein and increases the translation of selective
mRNAs (ribosomal proteins, translation elongation
factors, etc). Importantly, it is activated by most
of the hypertrophic agonists, such as angiotensin,
calcium, protein kinase C. Body size of the S6K 1
knockout mice is smaller than control.
Because rapamycin potently inhibits cellular growth
and cytokine production and regulates effectors of
the insulin pathway, rapamycin may effectively suppress
cardiac hypetrophy. To test this possibility, rapamycin
was given to adult mice with ascending aortic constriction.
Heart weight increased by 35%, mostly occurring within
48 hours after the operation. S6K activity was highest
4 hours after the operation and returned to nearly
normal at 48 hours. This result may be comparable
with the natural growth curve of the heart, because
growth of the heart stops at 48 hours. S6 phosphorylation
was highest 4 hours after the operation. Rapamycin
completely suppressed S6 kinase activation in the
binded heart 4 hours after the operation. S6 phosphorylation
was also completely suppressed.
Rapamycin did not induce lethality or body weight
loss in adult mice. Heart weight increased about 35%
in the banded mice and rapamycin significantly attenuated
the heart weight increase by 60%. On echocardiographic
examination, rapamycin significantly reduced the LV
end diastolic diameter. Rapamycin did not affect cardiac
contractility.
|
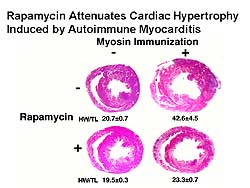 |
| Figure
2. Rapamycin attenuated cardiac hypertrophy in
a rat autoimmune myocarditis model. |
| Click
to enlarge |
|
To examine the effects of rapamycin on primary disease,
rapamycin was given to a rat autoimmune myocarditis
model. Cardiac myosin immunization into Lewis rats
induced autoimmune myocarditis. Three weeks after
immunization, extensive myocardial inflammation was
observed and heart weight was increased (Figure
2). Rapamycin almost completely suppressed the
increase in heart size.
In summary, mTOR appears to be a good candidate for
heart failure treatment. But several questions must
be resolved, including whether rapamycin can reverse
established cardiac hypetrophy and prevent or reverse
cardiac dilation. Despite the significant amount of
work to study the signaling mechanism of cardiac hypertrophy
and failure, a gap between basic information and clinical
practice remains. Methods to deliver drugs or genes
in a heart-specific manner to avoid systemic toxicity
are needed. Rapamycin target molecules will be useful
for improved treatment of heart failure.
|
PAGE
TOP
Therapeutic Myo-Angiogenesis for Ischemia-Induced Myocardial Remodeling by Transplantation of Autologous Bone Marrow Mononuclear Cells
Hiroaki Matsubara
Kansai Medical University, Moriguchi, Japan
|
|
The current concept of angiogenesis is that endothelial
progenitor cells in the peripheral circulation migrate
to the inflammatory areas and adhere and differentiate
into the endocardium. Bone marrow mononuclear cells
(BMMC) contain cardiac progenitor cells and release
angiogenic factors, such as bHGF and VEGF. Matsubara
and colleagues hypothesized that implanting BMMC into
ischemic limb or heart may enhance angiogenesis or
cardiomyogenesis. Culturing human marrow CD34+ cells
with human endothelial cells resulted in a network
formed with endothelial cells.
In the pig acute or chronic ischemia model, BMMC were
injected into the ischemic areas by open thoracotomy
and catheter-based injection. On coronary angiography,
collateral vessels were observed prior but not after
the injection. Neocapillary vessel formation was increased
about 2-fold after injection. The bone marrow cells
were differentiated in the endothelium. In a further
experiment, in which GFP-positive bone marrow stem
cells were injected into the scar area in the pig
model, there was little differentiation into cardiomyocytes
from the GFP lin-, cKit-, Sca-1 bone marrow hematopoietic
stem cells. Cell therapy using bone marrow cells results
in angiogenesis, not myogenesis.
This group has performed pre-clinical studies of marrow
cell implantation in the ischemic limb model in the
rabbit, acute myocardial infarction in the pig, and
in the chronic myocardial ischemic model in the pig.
These studies showed that there is no cardiac injury
by inflammatory cytokines released from implanted
bone marrow cells, no differentiation of other lineage
cells (osteoblasts, fibroblasts), and no malignant
arrhythmia on Holter monitoring. This shows the safety
and feasibility of bone marrow implantation into ischemic
myocardium. The cells should be injected into the
hibernating myocardium.
The Therapeutic Angiogenesis by Cell Transplantation
(TACT) clinical trial studied the effect of bone-marrow
derived cells implantation in patients with ischemic
heart disease (IHD) and peripheral artery disease
(PAD). This multicenter study in Asia and the US studied
96 patients.
The results of the Sole Cell Therapy Phase I arm of
this trial in patients with IHD were reviewed. NOGA™
3-D electromechanical mapping is used to map the hibernating
area for the catheter-based cell delivery. Voltage
and mechanical mapping was analyzed simultaneously.
Wall motion was improved at 6 weeks in the pig injected
with bone marrow cells compared to saline control
injection.
One patient with CCS class IV angina has been studied:
a 64-year-old male patient with an old MI with no
medical or therapeutic revascularization options and
who was using nitroglycerin spray 10-15 times per
day. On left coronary angiogram, before the implantation,
no blood vessels were seen around the left circumflex
and the injection was made in this area. This is the
first patient in the world to receive this type of
implantation, which was done using the open heart
method.
Spect-sestamibi scan showed improvement of myocardial
ischemia at 2 months. At 6 weeks, on NOGA mapping,
improvement was seen. The left ventricular ejection
fraction improved from 42% before the procedure to
53% at 6 weeks post-procedure. Chest pain disappeared
and cardiac function improved. The use of nitroglycerin
spray decreased from 10 times per day before the procedure
to 2 times in the 6 months and 6 times in the 12 months
after the procedure. The number of PVCs decreased
from 500-1000/day before the procedure to <100/day
at 6 months post-procedure. There were no side effects.
The second clinical case was a 59-year-old woman with
medically refractory angina (CCS 3-4), with diabetes,
ASO, hypertension, heart failure, and an ejection
fraction of 45%. She had a history of PCI and bypass
surgery. Catheter injection of bone marrow cells were
made into 20 different sites in the heart. At week
8 post-procedure, wall motion was improved. Spect-sestamibi
scan showed improvement of the distribution areas.
In summary, for future regeneration therapy for IHD,
angiogenesis is best for the ischemic portion. The
tools are bone marrow cells or angiogenic factors
(VEGF, bFGF, HGF). For the infarcted scar portion,
myocyte regeneration is needed, and the tools used
in the US and Europe are skeletal myoblasts. Presently,
autologous transplantation is prevalent. Another potential
cell is the cardiac stem cells, which may come from
the bone marrow or from cells originating from the
heart or skeletal myoblasts.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2003
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|