|
|
|
|
| Diagnosis and Treatment of Diastolic
Heart Failure: From Bench to Bedside |
|
|
iNOS Regulates Diastolic Dysfunction in the
Development of Heart Failure
Gen Takagi
Nippon Medical School-Chiba
Hokusoh Hospital, Chiba, Japan
Stabilization
of Calcium Release Channel (Ryanodine Receptor)
Is Involved in the Cardioprotective Mechanism
by Angiotensin II Receptor Blocker in Heart
Failure
Masafumi Yano
Yamaguchi University, Ube,
Japan
Extracellular Collagen Matrix Determines Left Ventricular Shape, Function and Stiffness during the Process of Ventricular Remodeling
Yasuki Kihara
Kyoto University Graduate School of Medicine, Kyoto, Japan
Extracellular
Matrix Remodeling as a Determinant of Transition
to Diastolic Heart Failure in Hypertensive Hearts:
Its Diagnostic and Therapeutic Approach
Kazuhiro Yamamoto
Osaka University Graduate
School of Medicine, Suita, Japan
Assessment
of Left Ventricular Diastolic Function Independent
of Cardiac Translation Using Newly Developed
Tissue Strain Imaging with Tissue Tracking Technique
Tomotsugu Tabata
University of Tokushima,
Tokushima, Japan
|
|
|
|
|
iNOS Regulates Diastolic Dysfunction in the
Development of Heart Failure
Gen Takagi
Nippon Medical School-Chiba Hokusoh
Hospital, Chiba, Japan
|
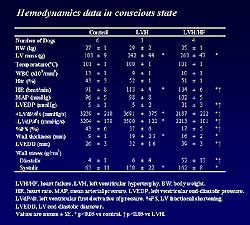 |
| Figure
1. Hemodynamic
data in the three study groups in the present study. |
| Click
to enlarge |
 |
| Figure
2. The
function of isolated myocytes in the three study groups. |
| Click
to enlarge |
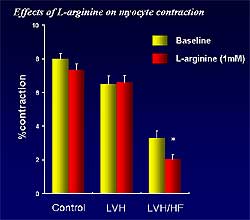 |
| Figure
3. Myocyte function is significantly reduced in heart failure, but not
hypertrophy or controls, by L-arginine, a nitric oxide synthase substrate. |
| Click
to enlarge |
|
Inducible nitric oxide (iNOS) plays a functional
role in the development of cardiac decompensation
from severely hypertrophied hearts, according to a
study presented by Takagi.
The mechanisms by which nitric oxide (NO) regulates
the failing heart have been unclear. The lack of understanding
may be due to differences in isoform expression and
function of NO, that is, iNOS, neuronal NOS, and endothelial
NOS may play different roles. Whether iNOS simply
plays a role in mediating cytokines, inflammation
and vascular function, or whether it exerts an action
on the function of the myocyte in heart failure, has
not been known.
The present study evaluated the role of iNOS in regulating
diastolic dysfunction in the development of left ventricular
hypertrophy (LVH) progressing to heart failure (HF)
in a canine model. The animals were evaluated in the
LVH stage and in the HF stage, and compared to control
canines (Figure
1).
The study demonstrated an important role for iNOS
with regard to myocyte function in heart failure (Figure
2). iNOS expression (protein level and localization)
was significantly enhanced in HF myocytes, and this
correlated with diastolic function. Expression of
iNOS was not enhanced in LVH or in control animals.
Isolated myocyte length was elongated significantly
in LVH and HF compared to controls. Myocyte systolic
function and diastolic function were significantly
depressed in LVH and HF, compared with controls. L-arginine,
a NOS substrate, significantly reduced myocyte function
in HF, but not in LVH or controls (Figure
3). Both a specific and a nonspecific iNOS inhibitor
abolished this effect of L-arginine in HF myocytes.
|
PAGE
TOP
|
Stabilization
of Calcium Release Channel (Ryanodine Receptor) Is
Involved in the Cardioprotective Mechanism by Angiotensin
II Receptor Blocker in Heart Failure
Masafumi Yano
Yamaguchi University, Ube, Japan
|
|
The angiotensin II receptor blocker valsartan corrected
the abnormal function of the sarcoplasmic reticulum
that occurs in heart failure in a study from Yamaguchi
University.
Abnormal function of the saroplasmic reticulum (SR)
is a major pathogenic mechanism in heart failure.
In a canine model of heart failure, valsartan treatment
did not improve hemodynamics, but corrected SR function,
Yano reported.
In heart failure, an abnormal Ca2+ leak occurs through
the ryanodine receptor (RyR). This is due to partial
loss of RyR-bound FKBP12.6 and the resulting conformational
change in the RyR. The investigators sought to determine
whether low-dose valsartan could correct the defective
interaction of FKBP12.6 and the RyR in experimental
heart failure.
|
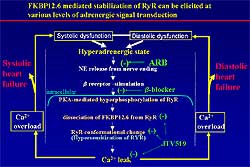 |
| Figure
1. FKBP-mediated stabilization can prevent the
development of heart failure, by interfering at
different levels of adrenergic signal transduction.
|
| Click
to enlarge |
|
An abnormal SR Ca2+ leak was found in untreated failing
SR, but no abnormality was observed in dogs treated
with valsartan. Valsartan treatment restored the stoichiometry
of the RyR versus FKBP12.6 that was decreased in untreated
SR. In the untreated group, the RyR was PKA-hyperphosphorylated,
whereas valsartan inhibited PKA hyperphosphorylation
of RyR and increased RyR-bound FKBP12.6. The amount
of the RyR-bound FKBP12.6 that was tremendously reduced
in the untreated group was reversed with valsartan.
Both SR Ca2+ uptake function and the amount of Ca2+-ATPase
were also decreased in untreated SR, whereas they
were restored with valsartan treatment. Valsartan
treatment did not improve left ventricular contractility
and relaxation at rest, but it did improve the contractile
response to dobutamine. Figure
1 illustrates how FKBP-mediated stabilization
can interfere and prevent heart failure.
Although valsartan did not improve cardiac function,
it corrected SR function. This apparently discordant
effect of valsartan may be due to its beta-blockade
type action.
|
PAGE
TOP
|
Extracellular
Collagen Matrix Determines Left Ventricular Shape,
Function and Stiffness during the Process of Ventricular
Remodeling
Yasuki Kihara
Kyoto University Graduate School
of Medicine, Kyoto, Japan
|
 |
| Figure
1. Factors involved in the dynamic regulation
of extracellular collagen matrix. |
| Click
to enlarge |
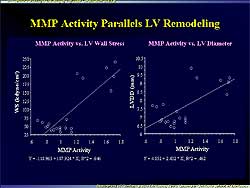 |
| Figure
2. Left ventricular wall stress and LV diastolic
diameter were increased in relation to MMP activation
in the present study. |
| Click
to enlarge |
|
The myocardial cell-to-cell connection through the
extracellular collagen matrix (ECM) is important in
maintaining left ventricular shape, contraction, and
stiffness (Figure
1). ECM may be degraded during the process of
left ventricular remodeling (LVR), primarily mediated
by the local fibroblasts. The balance between matrix
metalloproteinases (MMP) and the tissue inhibitors
of MMP (TIMP) appears to be critical for the downstream
degradation. Investigators at Kyoto University Graduate
School of Medicine found that this ECM breakdown may
occur through intrinsic MMPs.
Their study utilized Dahl salt-sensitive rats with
hypertension from the age of 11 weeks (compensated,
concentric hypertrophy/left ventricular hypertrophy
[LVH] stage) until 17 weeks (the LVR stage).
The rats were orally administered ONO-4817, a relatively
specific MMP-2 inhibitor, or vehicle.
Collagen type I and III were markedly upregulated
in the hypertrophy stage, but not in the heart failure
stage, in the study rats compared to age-matched normotensive
control rats. Zymography showed that both MMP-2 activity
and TIMP activity remained inactive in the hypertrophy
stage, however the activity of both began to be increased
in the heart failure stage. The activation begins
at the transcriptional levels.
In the control rats, no changes in MMP activity were
found in the hypertrophy stage, but in the remodeling
stage net MMP activity increased by 89.2%. This increase
was closely related to increases in LV diastolic diameter
and systolic wall stress and to a decrease in LV stiffness
(Figure
2). This confirms the upregulation of ECM during
hypertrophy, which returns towards baseline in heart
failure. ECM degradation remained inactive in hypertrophy,
but was de novo activated during heart failure transition.
Activity of ECM degradation paralleled the LV dilation
and mechanical decompensation.
|
|
In rats receiving ONO-4817, net MMP-2 activity was
suppressed by 27.4% and ECM appeared much denser.
In addition, rats receiving ONO-4817 had longer survival
than the control rats, about 25-27 weeks compared
to 20 weeks for control rats.
In vivo echocardiography showed that in control rats
LV diameter increased after 15 weeks, associated with
LV wall thinning, decreased LV fractional shortening,
and increased LV systolic wall stress, while in the
MMP inhibitors groups small LV size, LV shape, systolic
wall function, and normal LV wall stress were maintained.
Blood pressure levels were not affected by the MMP
inhibitor. Therefore, endogenous MMP inhibitor appears
to act along with intrinsic MMP activation to provide
sufficient suppression of its activation and prevent
LV remodeling. Scanning electron microscope showed
clear degradation of ECM in the heart failure stage,
compared to the hypertrophy stage, while treatment
with ONO-4817 showed recovery of the dense collagen
network.
In conclusion, an MMP inhibitor, ONO-4817, suppressed
the endogenous MMP activation and blocked the ECM
degradation that occurred during heart failure transition.
The preserved ECM maintained the LV shape, systolic
function, and diastolic properties, thus improving
animal survival. Pharmacological inhibition of ECM
degradation could be an adjuvant therapy for patients
undergoing ventricular remodeling.
|
PAGE
TOP
|
Extracellular
Matrix Remodeling as a Determinant of Transition to
Diastolic Heart Failure in Hypertensive Hearts: Its
Diagnostic and Therapeutic Approach
Kazuhiro Yamamoto
Osaka University Graduate
School of Medicine, Suita, Japan
|
|
Extracellular matrix (ECM) remodeling plays crucial
roles in diastolic heart failure (DHF), and because
of the close relation between fibrosis and brain natriuretic
peptide (BNP), the elevation of plasma BNP may be
a hallmark of patients with DHF.
Yamamoto and colleagues developed a hypertensive DHF
model using Dahl-Iwai salt-sensitive rats to evaluate
how myocardial stiffening induces DHF. Myocardial
stiffening was not promoted by ventricular hypertrophy
(LVH) but by ventricular fibrosis with enhanced collagen
cross-link and an increase in type 1, rather than
type III, collagen.
Further investigations revealed that BNP may be a
useful marker for this structural remodeling. In the
DHF model, BNP was associated with maladaptive LVH
with progressive ventricular fibrosis, but was not
associated with compensatory LVH with subtle fibrosis.
This suggested that BNP may be able to discriminate
patients at risk for DHF. Indeed, in clinical studies,
patients with a history of acute pulmonary edema due
to DHF were shown to have higher plasma BNP levels
than asymptomatic hypertensive patients with similar
ventricular mass, Yamamoto said.
In the DHF model, progression of ventricular fibrosis
was associated with phospholipase D (PLD) activation
that was induced by growth factors and agonists binding
to G-protein-coupled receptors. Since ethanolamine
is a product of PLD, the researchers hypothesized
that N-methylethanolamine, an analogue of ethanolamine,
would decrease PLD activity through a negative feedback
mechanism, suppressing collagen production and preventing
myocardial stiffening. Indeed this was the case, as
this agent suppressed PLD activity and both mRNA and
protein levels of collagen without depressor effects,
and prevented stiffening. N-methylethanolamine, therefore,
may exert therapeutic effects on ventricular fibrosis
by inhibiting PLD, independent of stress unloading.
|
PAGE
TOP
Assessment
of Left Ventricular Diastolic Function Independent
of Cardiac Translation Using Newly Developed Tissue
Strain Imaging with Tissue Tracking Technique
Tomotsugu Tabata
University of Tokushima, Tokushima,
Japan
|
|
Tissue strain imaging with region-of-interest (ROI)
tracking can potentially evaluate left ventricular
(LV) diastolic function independent of preload and
cardiac translation, according to Tabata, who, with
his colleagues, developed the new technique.
LV diastolic function cannot be consistently evaluated
by the Doppler transmitral inflow velocity pattern
because of its preload dependency. The pulsed tissue
Doppler mitral annular motion velocity pattern (TDI)
evaluates LV diastolic function believed to be relatively
preload-independent, but is limited by the effect
of cardiac translation. To overcome the problem of
cardiac translation, tissue strain imaging (TSI) was
recently developed using color TDI technique (ApliQ,
Toshiba Corp.).
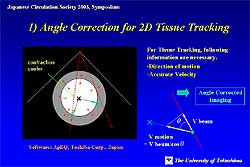
With this technique, the center of contraction was
set in the LV cavity and velocity was automatically
angle-corrected (Figure
1). The velocity values from the same region of
moving myocardium were automatically defined and interrogated
over time to yield displacement by 2D tissue Doppler
tracking technique. TSI was finally obtained as a
spatial derivative of the tissue displacement.
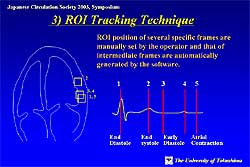
A study was performed to evaluate LV diastolic function
using variables obtained by TSI with ROI tracking
(Figure
2). The study evaluated longitudinal strain rate
in 20 normal hearts, 35 hypertrophied hearts and 8
hearts with dilated cardiomyopathy (Figure
3).
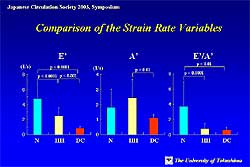
The experiments showed that the ratio of early diastolic
strain rate obtained from TSI to transmitral E wave
velocity consistently decreased from normal to pseudonormal.
With this technique, the difference between normal
hearts and hypertrophied hearts was highly significant
(P <0.0001), as was the difference between normal
hearts and those with dilated cardiomyopathy (P <0.0001)
(Figure
4). The difference between hypertrophied hearts
and those with cardiomyopathy was significant at P
<0 .01.
With other techniques, these differences were not
revealed. For example, a comparison of the velocity
variables by pulsed tissue Doppler showed early diastolic
wave to be reduced in hypertrophied hearts and in
hearts with cardiomyopathy, versus normal, but the
two abnormalities appeared similar and could not be
differentiated from each other.
The investigators concluded that the TSI potentially
evaluates left ventricular diastolic function in a
manner that is free from the problems of preload dependency,
cardiac translation, Doppler angle, and tissue tracking.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2003
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|