|
|
|
|
| Molecular Mechanisms of Heart Failure |
|
|
Calcium Signaling and Heart Failure: Defective Inter-Domain Interaction within the Ryanodine Receptor
Masafumi Yano
Yamaguchi University School of Medicine, Yamaguchi, Japan
Telomerase, Cyclins, and Cardiac Apoptosis
Michael D. Schneider
Baylor College of Medicine, Houston, TX
The Role of the Mitogen-Activated Protein Kinase Family in the Transition to Heart Failure
Kinya Otsu
Osaka University Graduate School of Medicine, Osaka, Japan
Role of Oxidative Stress in Post-Infarct Left Ventricular Remodeling and Failure
Hiroyuki Tsutsui
Hokkaido University Graduate School of Medicine, Sapporo, Japan
|
|
|
|
|
Calcium Signaling and Heart Failure: Defective Inter-Domain Interaction within the Ryanodine Receptor
Masafumi Yano
Yamaguchi University School of
Medicine, Yamaguchi, Japan
|
|
Work by Yano and colleagues showed that the specific
domain interaction with the ryanodine receptor (RyR)
critically regulates the gating property of the RyR,
and that its defectiveness may be involved in the
common pathogenic mechanism of heart failure. The
restoration of the defective domain interaction may
provide a new clue for the development of a therapeutic
strategy for heart failure.
|
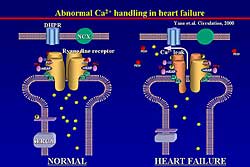 |
| Figure
1. Schematic of abnormal calcium handling in normal
setting and heart failure. |
| Click
to enlarge |
|
The RyR displays a micro-molecular complex, which
includes 4 identical subunits and a variety of excessive
proteins, such as phosphatase, PKA, and FK binding
protein (FKBP). FKBP is known to stabilize channel
gating with 1 RyR to 4 FKBP. In normal conditions,
the channel is stabilized, but in heart failure, as
this group reported previously, 3 of the 4 FKBPs are
dissociated from the RyR and a comfirmation change
of the RyR occurs and abnormal calcium leak is induced
(Figure
1).
The aberrant calcium leak is at the level of a single
channel. In heart failure, channel gating is
hypersensitized to calcium: at a lower concentration
of calcium, the channel is more activated. This can
be seen in malignant hyperthermia, known as single
point mutation disease of the skeletal type of RyR.
|
|
Many mutation sites have been reported for malignant
hyperthermia or central core disease. Interestingly,
these mutation sites cluster in 3 regions: N-terminal
domain, central domain, and channel forming domain.
Corresponding with these 3 regions, several mutations
have been reported recently in patients with arrhythmogenic
right ventricular dysplasia (ARVD) or polymorphic
ventricular tachycardia (pVT). Because single point
mutation at any region critically changes the channel
gating, these regions are considered to be very important
for the regression of channel gating.
Interestingly, the FKBP binding site is very closely
located to the central region. Other mutations at
the N-terminal domain or central domain induce hypersensitization
of the channel. Ikemoto and colleagues proved their
hypothesis, using skeletal type RyR, that there is
an interaction between the N-terminal and the central
domain as a regulatory switch for channel gating.
|
|
Study design and results
In the present study, Yano and colleagues assessed
whether the abnormality in the inter-domain interaction
induces the defective channel gating of RyR in heart
failure. Under normal conditions, the N-terminal domain
and the central domain interact, and then the channel
is stabilized. The mode of the regulatory domain is
called zipping.
They hypothesized that a mutation at either the N-terminal
or the central domain interferes with the domain interaction
and results in unzipping, which may de-stabilize the
channel.
The domain peptide approach was used to prove their
hypothesis. The domain peptide named DPc10 was used,
which is a copy of the specific region of the central
domain, has about 40 amino acid residues, and contains
the reported mutation site. When DPc10 is introduced
it is supposed to bind in the terminal region, and
then in competition with native amino acid residue,
unzipping may occur.
They confirmed that DPc10 binds to the RyR, as assessed
by site-directed fluorescent labeling using the fluorescence
confirmation probe SAED. Recently, in other work,
they confirmed the fluorescence is located in the
internal region. So, DPc10 binds to the N-terminal
region.
To quantitate the mode of the domain interaction,
they performed a chemical quencher experiment. In
the unzipping state, a large chemical quencher, such
as QSY-BSA, can easily access the gap between the
regulatory domains and then efficiently quench the
fluorescence. But, in the zipping state, the large
chemical quencher cannot access the gap of the regulatory
domain and the fluorescence cannot be quenched. In
the presence of DPc10, the quenching extent was increased,
indicating the shift of the mode from zipping to unzipping.
FK506 also induced a shift of the mode from zipping
to unzipping.
DPc10 dose-dependently induced calcium leak from
the sarcoplasmic reticulum (SR). ATP was added, causing
calcium uptake, and then after its completion, the
calcium ATPase blocker thapsigargin was added to visualize
the subsequent calcium leak. In the absence of DPc10,
there was little calcium leak, but the leak appeared
with an increase in the dose of DPc10.
DPc10 decreased the RyR-bound FKBP12.6, although
DPc10 had no effect on the level of the phosphorylation
of the RyR. Thus, this demonstrated that FKBP can
be dissociated from the RyR, even without a change
in the level of phosphorylation of the RyR.
Based on these findings, it is likely that FKBP dissociation
induces domain unzipping and also that domain unzipping
induces FKBP dissociation, as a regulating mechanism
for channel gating.
Next, the effect of DPc10 on the in vivo myocyte
function was assessed. After delivering DPc10 into
the cell, calciumtransient and cell shortening were
measured using Fura 2. In normal myocytes, in response
to forskolin, an adenyl cyclase activator, cell shortening
and the calcium transient were increased. In the presence
of Dpc10, cell shortening and the peak of the calcium
transient were decreased, the duration of the calcium
transient was prolonged, and the response to forskolin
was very poor. However, when incubating the myocyte
with the calcium channel stabilizer JTV519, cell shortening
and calcium transient were restored and the response
to forskolin was restored toward normal. Previously
they reported that JTV519 restores the confirmation
state of the RyR and prevents abnormal calcium leak
through the RyR.
Because this is similar to what is seen in the failing
myocyte, they measured cell shortening in the failing
myocyte taken from the pacing-induced canine heart
failure. In failing myocytes, cell shortening and
calcium transient were deteriorated, and the response
to forskolin was very poor. But, after incubating
the failing myocytes in JTV519 for 12 hours, cell
shortening and calcium transient were restored, and
the response to forskolin was restored towards normal.
The regulatory domains in the failing myocytes were
then evaluated. Interestingly, as in the case in which
they introduced DPc10 in the normal myocyte, in the
failing myocyte unzipping already occurred. The RyR
is hyper-phosphorylated in association with a decreased
amount of RyR-bound FKBP, and spontaneous calcium
leak occurs. But in the presence of the channel stabilizer
JTV519, the mode of the domain interaction shifted
from the unzipping state to the zipping state. Spontaneous
calcium leak disappeared.
|
|
Conclusion
Excessive beta-receptor stimulation induces PKA-mediated
hyper-phosphorylation of the RyR, which induces the
dissociation of FKBP. This may shift the mode of the
domain interaction from zipping to unzipping. This
domain zipping may lead to calcium leak and intracellular
diastolic calcium overload, systolic and diastolic
dysfunction, arrhythmia, and finally the development
of heart failure. The mutations seen in AVRD or pVT
directly induce the shift of the domain interaction
from zipping to unzipping.
A beta blocker can prevent the abnormal flow at the
beta-receptor level. JTV519 may directly affect the
RyR and shift the mode of domain interaction from
zipping to unzipping. JTV519 may inhibit domain unzipping.
|
PAGE
TOP
|
Telomerase,
Cyclins, and Cardiac Apoptosis
Michael D. Schneider
Baylor College of Medicine, Houston,
TX
|
|
This lecture focused on the cytoprotective roles
of telomerase and what has been learned about how
telomere dysfunction kills cardiac myocytes.
The extrinsic apoptotic pathway involves death domain
receptors and the intrinsic apoptotic pathway or mitochondrial-dependent
pathway is triggered by familiar cardiovascular (CV)
stressors such as oxidative stress and IGF receptors
working AKT. Based on increasing evidence for the
prevalence of apoptosis in acute myocardial damage
in chronic heart failure states, investigators have
sought to manipulate this cascade in a manner favorable
for survival of cardiac myocytes. For example, by
transgenic overexpression of Bcl2 family proteins
or pharmacological manipulations with caspase inhibitors.
However, particularly for long-term heart failure,
the adverse effects of interfering with death surveillance
pathways in bone marrow and other systems may be problematic.
Schneider and colleagues have chosen to focus primarily
in the initiating signals for cardiac myocyte apoptosis,
and have learned some new and interesting insights
into how conventional CV stresses are transduced into
apoptotic signals. Their work with 2 unpublished pathways
is reviewed here: 1) telomere dysfunction through
the activation of a proximal MAP kinase pathway, whose
key players are HGK (MAP4K4) and TAK1 (MAP3K7. These
are upstream activators of the JNKp38 pathways and
lead to cell death via loss of mitochondrial potential.
2) the involvement of PAL-2 directed cyclin-dependent
kinases, in particular, cyclin-dependent kinase 9
(T/cdka), that leads to mitochondrial dysfunction
through the suppression of a key transcriptional coactivator
for the expression of mitochondrial genes.
|
|
Telomere dysfunction
Telomeres are the DNA protein structure existing
at the specialized ends of chromosomes and cap the
linear ends of linear genomes. Telomerase reverse
transcriptase (TERT) is an RNA-dependent DNA polymerase,
using a specific RNA template. TERT maintains the
telomeric repeat, preventing telomere erosion and
“uncapping.” In adults, TERT is associated
primarily with germ cells, tumor cells, and stem cells.
TERT is markedly downregulated in myocardium after
birth. Transgenic mice were made that expressed wild-type
TERT or catalytically inactive TERT under the control
of cardiac specific alpha-myosin heavy chain (HMC)
promoter. In early hyperplasia, a larger than normal
number of smaller than normal of cardiac myocytes
were found. They also demonstrated that forced expression
of telomerase was cytoprotective for the heart. TUNEL-positive
cells post-infarction were reduced by 50%, with a
slightly smaller decrease in infarct size, normalized
for the area of risk. TERT also promotes myocyte survival
after mechanical stress. In mice subjected to severe
aortic banding, in the group with the greatest degree
of constriction, TUNEL-positive cells, interstitial
fibrosis, and systolic dysfunction are seen after
just 1 week of load. Forced expression of telomerase
prevents all 3 of those adverse aspects.
The biochemical consequences following mechanical
load are telomere erosion within just 1 week and the
transgene increases the basal telomere length and
prevents its loss. This was accompanied by partial
loss of 1 of those telomere repeating binding factors
(TRF-2) and activation of known TRF-2 dependent responses,
notably the phosphorylation and increased activity
of the DNA damage checkpoint kinase, known as Chk2.
All 3 of these responses were also seen in adult heart
failure patients: telomere erosion, loss of TRF-2,
and ATM-dependent phosphorylation of Chk2. Importantly,
in age-matched patients with hypertrophic cardiomyopathy
undergoing septal ablation surgery, and found neither
apoptosis, telomere erosion, or the biochemical responses
of the telomere were seen.
Studies for the mediators for the loss of TRF-2 and
its ability to confer the apoptotic state led them
to TGF-beta activated kinase 1, MAP3K7, and one of
its upstream activators HGK (MAP4K4). TAK1 was originally
described as an essential mediator of the BMP/TGF-beta
cascade. Komuro and colleagues have shown that TAK1
acts in a divergent-convergent relationship with respect
to the Smad transcription factors that are directly
phosphorylated by these receptors. Further, they have
shown an essential role for TAK1 in cardiac muscle
specification. But, TAK1 also occupies a central role
in signal transduction by other cascades, including
cytokines and IRAKs. TAK1 also has a complex role
as a negative regulator of the conical Wnt pathway.
TAK1 activation by cytokines is thought to involve
the upstream MAP kinase MAP4K4, which is a heart-enriched
protein kinase for which little or no functional information
was available.
Because existing antibodies to HGK were insufficient
for their purposes, they used transgenic mice expressing
epitope-tagged HGK in the heart. A number of CV stresses
activate HGK in an immune complex assay, including
transgenic expression of TNF-alpha and the G protein
Gq, the essential signaling intermediate for angiotensin
II, endothelin, and is an intermediate for mechanical
signal transduction in the heart. Similar activation
was seen by ischemia, reperfusion injury, and mechanical
load in the epitope-tagged HGK mice.
Although HGK induced no discernible phenotype in
cardiac muscle at baseline, it had a marked adverse
synergy in relation to Gq. The Gq mice had mild concentric
hypertrophy, and a very slight but significant increase
apoptotic cells. The double transgenic mice had severely
enlarged hearts with dilated cardiomyopathy (DCM),
a marked increase in apoptosis measured by TUNEL staining
and caspase activation, and severe early mortality.
They showed that a variety of stresses, including
Gq, TNF-alpha, oxidative stress, and ceramide, result
in downregulation of TRF-2, the essential telomere
capping protein. The stress-induced downregulation
of TRF-2 is occurring through the HGK-TAK1 module.
The loss of TRF2 in ceramide-treated cells is partially
rescued by dominant-negative HGK or dominant-negative
TAK-1, with a larger rescue imposed by Bcl2, a conical
apoptotic protein. So, stress pathways downregulate
TRF2 via HGK-TAK1. Conversely, the loss of TRF2 function
or expression activates HGK. This was shown using
wild-type versus dominant-negative TRF2 or in antisense
knockdown of the endogenous of TRF2.
To summarize the role of telomerase in cardiac protection,
endogenous telomerase is downregulated in adult myocardium.
Telomerase dysfunction is a consequence of cardiac
stress cascades in tissue culture, in animal models,
and in human heart failure.
A TRF2-MAP4K4 cycle amplifies apoptotic signals.
Stress leading to activation of HGK, TAK1, and JNK
lead independently to the loss of TRF2, and the circular
action of these signaling molecules amplifies apoptotic
signals. Exogenous telomerase prolongs cardiac myocyte
cycling, and confers resistance to stress. One prediction
from this model is that preventing the loss of TRF2
should be cytoprotective in myocardium. Recently they
have confirmed this prediction. TRF2 was overexpressed
under the control of the alpha-MHC promoter in doxirubicin-induced
cardiomyopathy and showed the delay in mortality imposed
by exogenous TRF2 and the significant rescue of cardiac
myocytes from apoptosis. Conversely, they expressed
the dominant-negative form of TRF2 in transgenic mice,
which induces spontaneous cardiac myocyte apoptosis
at a low level and spontaneous cardiomyopathy with
onset at 8-9 months. Thus, they believe this is an
essential pathway for cardiac myocyte protection and
it is also a pathway that has therapeutic implications.
|
|
PAL2 directed cyclin-dependent kinases and
mitochondrial dysfunction
The important role of atypical cyclin-dependent kinases
(Cdk7, Cdk9) in cardiac myocyte hypertrophy was recently
described by Sano and colleagues. The substrate for
Cdk7 and Cdk9 is the large subunit of RNA polymerase
II in a highly multimerized, serine-rich carboxy terminal
domain (CTD). The unphosphorylated form of PAL2 is
recruited to promoters, along with the basal transcription
factors. Cyclin H and Cdk7 correspond to basal transcription
factor TF2H and are important for PAL2 to engage splicing
factors and execute pre-mRNA processing.
However, an additional phosphorylation is required
for PAL2 to leave the promoter proximal region and
enter the open reading frame, which occurs through
Cdk9. This complex is also known as positive transcription
elongation factor B (P-TEFb).
Both Cdk7 and Cdk9 are activated in vivo by hypertrophic
signals, including Gq, calcineurin, and long-term
mechanical load, in mice, as shown by Muroaki and
colleagues. Cyclin T was expressed under the control
of the alpha-MHC promoter and found a dose-dependent
increase in Cdk9 activity, with concentric hypertrophy
and myocycte enlargement at the higher levels. On
the basis of these experiments, it might be inferred
that activation of hypertrophic growth through Cdk9
might be potentially useful as a way to promote myocyte
muscle restoration. However, that turned out not to
be true.
The questions prompted by this work for RNA-polymerase
II-directed cyclin-dependent kinases in heart failure
are: Is hyperphosphorylation of RNA polymerase II
germane to human heart failure, as extrapolated from
mouse models?
Does hyperphosphorylation of RNAPII interact functionally
with other hypertrophic pathways?
If synergistic or adverse effects exist, what are
the mediators?
Phosphorylation of endogenous PAL2 on the preferred
site by Cdk7, the preferred site by Cdk9, and immune
complex kinase activity of both kinases are characteristic
of human heart failure. In transgenic mice, the increase
in endogenous Cdk9 activity can be seen conferred
by the signaling protein Gq, with a somewhat greater
increase induced by transgenic expression of cyclin
T1. There is a synergistic effect on Cdk9 activity.
The combined inheritance of Gq at doses that induce
mild concentric hypertrophy plus cyclin T1 against
mild concentric hypertrophy induces a florid DCM with
extensive tissue fibrosis and apoptosis and early
mortality. Microarray studies showed that even in
the cyclin T1 mice that have little or no adverse
phenotype, there was already a marked dysregulation
of mitochondrial gene expression, including metabolic
enzymes and mitochondrial transcription factors. One
potential explanation for this generalized dysregulation
of the mitochondrial program was the marked suppression
for an essential coactivator of mitochondrial biogenesis,
the PPAR gamma coactivator 1 (PGC-1).
Even in the cyclin T1 mice in the absence of overexpression
of Gq, the mitochondrial had markedly diminished cristae
and a decreased level of organization. Many of the
mitochrondrial enzyme activities were depressed. By
adenoviral delivery of cyclin T plus Cdk9 in tissue
culture, they were able to test the functional consequences
of this overexpression. They found that Cdk9 activity
at physiological levels produced pronounced apoptosis
and mitochondrial gene dysregulation. Repression of
PGC-1 was a very early response to cyclin T plus Cdk9
in tissue culture. Maximal reduction of PGC-1 was
achieved at 24 hours, whereas the other mitochondrial
proteins continued to diminish over time. Cyclin T
plus Cdk9 was also sufficient to suppress SERCA2 and
certain other cardiac proteins. Importantly, forced
expression of PGC1 by itself is sufficient to rescue
cardiac myocytes from Cdk9-induced gene dysregulation.
The suppression of Cox5b and the mitochondrial transcription
factor Tfam and cardiac myocyte apoptosis were rescued
by PGC-1 to baseline levels.
They currently postulate that hypertrophic signals
that result in Cdk9 activation lead through a pathway
that is not yet understood to marked suppression of
PGC-1, a master regulator of mitochondrial biogenesis
and function. The known partners for PGC-1 include
PPAR gamma, nuclear respiratory factor, and MEF2.
Downregulation has been seen of genes that are known
targets for each of these dimers. This leads to marked
suppression of genes required for mitochondrial metabolism
and function, decreased resistance to apoptosis, and
predisposition to heart failure in vivo.
To summarize, Cdk9 activation and hyper-phosphorylation
of PAL2 are characteristic of mouse models, cultured
cells, and human heart failure Benign levels of Cdk9
activity, which are well tolerated by themselves,
cause florid heart failure when combined with other
hypertrophic signals. Excess Cdk9 activity leads to
global downregulation of genes for mitochondrial function,
including its master regulator PGC-1. These levels
repress PGC-1 acutely in culture, cause defective
expression of genes for mitochondrial function in
culture, and predispose to apoptosis. Restoring PGC-1
rescues cells from the adverse effects of Cdk9, and
suggests that PGC-1 may be a useful therapeutic target
in heart failure studies.
|
PAGE
TOP
|
The
Role of the Mitogen-Activated Protein Kinase Family
in the Transition to Heart Failure
Kinya Otsu
Osaka University Graduate School
of Medicine, Osaka, Japan
|
|
In their work with stress-activated MAP kinases,
such as the ASK1 and p38 signaling pathways in the
transition to heart failure, this group showed that
mechanical stress, such as pressure overload or ischemic
insult, activates the ASK1-JNK signaling pathway,
leading to left ventricular (LV) remodeling and heart
failure. P38 plays a protective role in the stress
response. Thus, the balance of JNK and p38 may determine
cell survival or cell death.
|
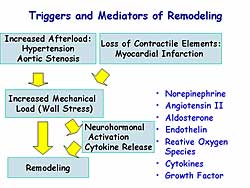 |
| Figure
2. The influence of triggers and mediators on
remodeling. |
| Click
to enlarge |
|
Cardiac remodeling, including changes to the geometry,
mass, volume, and function of the myocardium, is an
adaptive response manifested as cardiac hypertrophy.
The stimuli are continuous, excessive, and pathological,
such as hypertension, infarction, and inflammation.
The process can be maladaptive and the heart becomes
dilated, resulting in dysfunction, necrosis, and apoptosis.
Clinical symptoms of heart failure, such as congestion
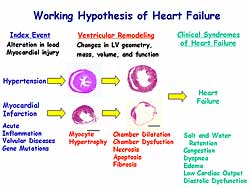
and dyspnea, then appear. Figure
1 summarizes the working hypothesis of heart failure.
Extensive research has been conducted to elucidate
the molecular mechanisms of cardiac remodeling. However,
more remains to be elucidated. The trigger for cardiac
remodeling is increased mechanical stress, by increased
afterload or loss of contractile elements (Figure
2). Neurohormonal activation and cytokine release
also are involved in this process.
Otsu and colleagues previously reported that apoptosis
signal regulating kinase 1 (ASK1) is activated by
angiotensin II, endothelin-I, TNF-alpha, and reactive
oxygen species (ROS), which are known to be a mediator
of cardiac remodeling. Thus, it can be hypothesized
that ASK1 may play a role in cardiac remodeling. Their
previous work showed that ASK1 is a ubiquitously expressed
MAP3 kinase, an upstream kinase that activates MKK4/7-JNK
and the MKK3/6-p38 signaling cascades. ASK1 plays
a role in the mechanism of stress-induced apoptosis.
|
|
Study with ASK1 knockout mice and TAC
To elucidate the in vivo role of ASK1 in cardiac
remodeling, this group used ASKI knockout mice (ASKKO).
Two experimental models of cardiac remodeling were
used: 1) a pressure overload-induced model, using
thoracic transverse aortic constriction- (TAC) induced
cardiac hypertrophy, with hypertrophy visible at 1
week and heart failure at 4 weeks; and 2) a myocardial
infarction (MI) model, created by ligation of the
left coronary artery. ASK1 activation was induced
by TAC and MI, as shown by in vitro assay.
The baseline parameters of the ASKKO and wild-type
(WT) mice showed no significant differences in body
weight, heart weight, and hemodynamic parameters.
The embryonic development and birth of the ASKKO was
normal and they were indistinguishable in appearance
from WT mice.
Surprisingly, although TAC induced an increase in
heart weight, cross-sectional area in the tissue section,
cell surface area in isolated cardiac myocyte, and
heart weight/body weight ratio, there was no difference
in these changes between the ASKKO and WT mice.
|
|
|
This group had previously reported that ASK1 plays
a role in cardiac hypertrophy with an in vitro system.
That data showed that angiotensin II, endothelin,
and PE are induced in the cardiac hypertrophic response,
but the dominant-negative form of ASK1 inhibited this
response. In contrast, constitutively active ASK1
can induce the hypertrophic response, suggesting that
ASK1 plays a role in cardiac hypertrophy. The precise
mechanism to explain this discrepancy is not known,
but one possibility is that the signaling pathways
leading to hypertrophy are multifold and one signaling
molecule is insufficient to prevent pressure-overload
hypertrophy.
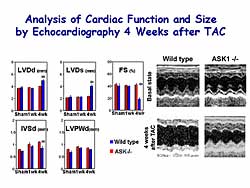
Pathological analysis at 4 weeks after TAC showed
that the heart dilation with fibrosis seen in the
WT was completely prevented in the ASKKO mice. Functional
analysis at 4 weeks after TAC showed that the WT mice
hearts are dilated and percent fractional shortening
was significantly decreased, but in the ASKKO mice
the heart dilation and decrease in percent fractional
shortening was prevented (Figure
3). This suggests that ASKI 1 may play a role
in cardiac remodeling by pressure overload.
|
|
Study with ASKKO mice and MI
|
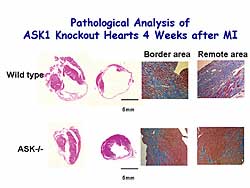 |
| Figure
4. Pathological analysis of ASK1 knockout mice
4 weeks after MI. |
| Click
to enlarge |
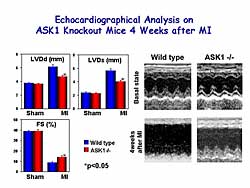 |
| Figure
5. Echocardiographic findings of ASK1 knockout
mice 4 weeks after MI. |
| Click
to enlarge |
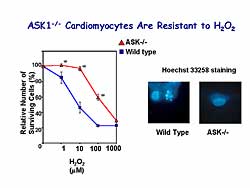 |
| Figure
6. ASK-/- cardiomyocytes are resistant to hydrogen
peroxide. |
| Click
to enlarge |
|
In this model, 4 weeks post-MI, the infarct area
was extended to the free wall, apex, and some of the
septum in the WT mice, and fibrosis replaced the ischemia
injury in the remote area. However, in ASKKO the infarct
was limited to the initial ischemic insult region
and the remote area appeared to be normal (Figure
4).
Pathological analysis 4 weeks post- MI revealed that
LVDd, LVDs, and percent fractional shortening were
increased in WT mice, but the cardiac remodeling was
significantly prevented in the ASKKO mice (Figure
5).
These data suggest that ASK1 plays an important role
in cardiac remodeling in pressure overload and in
MI. ASK1 is involved in stress-induced apoptosis.
At 1 week and 4 weeks after TAC, a large increase
in TUNEL-positive cells was seen, but this increase
was significantly attenuated in the ASKKO mice. The
TUNEL-positive cells are alpha-sarcomeric actin positive
cells, suggesting that an apoptotic cell is a cardiac
myocyte. These same findings were seen in the MI model.
There was an increase in apoptosis at 1 week and 4
weeks in the border and remote areas, but this increase
was significantly inhibited in the ASKKO mice in both
areas.
To confirm the involvement of ASK1 in apoptosis,
they isolated cardiac myocytes from the ASKKO mice.
An increase in hydrogen peroxide concentration was
associated with a decrease in the number of surviving
cells. However, ASKKO mice showed greater resistance
to hydrogen peroxide (Figure
6).
Constitutively active ASK1 can induce cardiac hypertrophy.
After infection of rat neonatal cardiomyocytes with
a higher titre of constitutively active ASK1, apoptosis
was induced, indicating that the extent of ASK1 activation
may determine the direction toward cardiac hypertrophy
or apoptosis.
|
|
Cardiac-specific knockout mice experiments
|
|
|
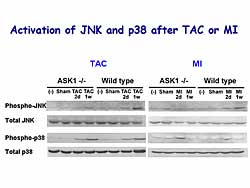
After TAC or MI, p38 is activated in WT mice, but
there was no significant difference in the activation
level in the ASKKO mice. However, there was significant
activation of JNK in WT mice after TAC and MI, but
this activation was significantly attenuated in ASKKO
mice (Figure
7). So, the ASK1-JNK signaling pathway may lead
to LV remodeling and heart failure. Apoptosis may
be involved in this process.
To elucidate the in vivo role of p38 in the stress
response, they generated p38 floxed allele mice, with
a functional exon containing the ATP binding site,
and they generated mice overexpressing Cre recombinase
under the control of the alpha myosin heavy chain
(MHC) promoter. Cross-breeding these mice resulted
in a cardiac-specific p38 KO mice, with a 90% reduction
of p38 alpha, without any significant differences
in the p38 isoform or MAP kinase.
|
|
No significant difference was found in the microscopic
or macroscopic appearance in the p38KO mice heart.
No significant difference was found in the function
or structure of the heart. So, there was no requirement
for p38 alpha during embryonic development. No significant
difference was seen in any of the measured physiological
parameters in p38KO mice.
At 1 week after TAC, there was no significant difference
in the hypertrophic response between the WT and p38KO
mice, such as LV weight, cross-sectional area, or
biochemical markers. This suggests that p38 does not
have an essential role in cardiac hypertrophy by pressure
overload. However, the p38KO mice hearts became dilated
with massive fibrosis, suggesting that p38 plays a
protective role in the stress response.
In the p38KO mice, 1 week after TAC, an increase
in LVd and LVds was found, and a decrease in percent
fractional shortening. In the p38KO mice there was
a large increase in the TUNEL-positive cells, which
were alpha-sarcomeric actin-positive, indicating they
are cardiomyocytes. An increase in the release of
cytochrome C into the cytosol was detected, as well
as an increase in the Bax/Bcl ratio, suggesting mitochondrial
pathways are involved in apoptosis in p38KO hearts.
|
PAGE
TOP
|
Role
of Oxidative Stress in Post-Infarct Left Ventricular
Remodeling and Failure
Hiroyuki Tsutsui
Hokkaido University Graduate
School of Medicine, Sapporo, Japan
|
|
Studies in transgenic mice by this group showed that
mitochondrial antioxidants, including glutathione
peroxidase (GSHPx) and peroxiredoxin-3 (Prx-3), and
mitochondrial transcription factor A (mtTFA) are novel
therapeutic target genes in heart failure. MtTFA is
important in the replication and maintenance of mitochondrial
DNA.
|
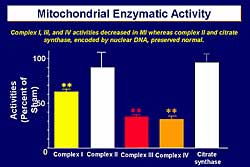 |
| Figure
2. Mitochondrial enzymatic activity in an experiment
in the post-infarct mouse model. |
| Click
to enlarge |
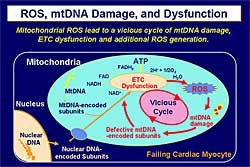 |
| Figure
3. The pathophysiology of reactive oxygen species,
mitochondrial DNA and dysfunction, based on experiments
by Tsutsui and colleagues. |
| Click
to enlarge |
|
Myocardial infarction is the most important cause
of heart failure. Post-infarct myocardial remodeling
plays an important role in the pathophysiology of
heart failure, and includes infarct expansion that
occurs hours to days post-infarct and the subsequent
global remodeling process that occurs days to months
post-infarct. Activation of the renin-angiotensin
system (RAS) in myocardial remodeling has led to the
use of ACE inhibitors as the first-line drug for the
treatment of the acute and chronic phases of heart
failure. The 23% risk reduction in mortality with
ACE inhibitors found in the SOLVD treatment trial
is insufficient. Elucidation is necessary of novel
contributing factors that must be downstream of neurohormones
that are activated in the setting of heart failure
and upstream of myocardial remodeling, which includes
hypertrophy, fibrosis, and apoptosis (Figure
1).
Thus, oxidative stress in the pathophysiology of
heart failure has been the focus of research by Tsutsui
and colleagues. Previous work by this group showed
that TNF-alpha and angiotensin II can generate reactive
oxidant species (ROS) in cardiac myocytes. In the
post-infarct mouse model they showed that 1) mitochondrial
superoxide generation was increased in the failing
heart, 2) mitochondrial DNA injury can be observed,
and 3) the mitochondrial copy number was significantly
decreased with a parallel decrease in mitochondrial
DNA-derived transcript messages encoding the mitochondrial
electron transport complexes, leading to a decrease
in the activity of Complex I, III, and IV (Figure
2). However, Complex II and citrate synthase are
encoded by nuclear DNA and were preserved at normal
levels.
Based on these results, mitochrondrial ROS leads
to mitochondrial DNA damage and electron transport
gene dysfunction, which then leads to additional ROS
generation—a vicious cycle between mitochondrial
damage and mitochondrial dysfunction (Figure
3). Therefore, the modulation of mitochondrial
oxidative stress can be a novel therapeutic strategy
for heart failure.
|
|
Study with GSHPx in transgenic mice \
GSHPx is a key antioxidant enzyme, which scavenges
hydrogen peroxide (H2O2) and prevents the formation
of other more toxic radicals such as the hydroxy radical
(OH). GSHPx possesses a higher affinity for H2O2 than
catalase, also a scavenger for H2O2 and OH. GSHPx
is present in high amounts within the heart, especially
in the cytosolic and mitochondrial compartments. Therefore,
GSHPx can exert greater protective effects against
oxidative damage than SOD, catalase, or their combination.
Myocardial infarction was created in GSHPx transgenic
mice (C57BL/6xCBA/J hybrid mice overexpressing GSHPx;
TG+MI group, n=44) and in wild-type mice (WT+M group,
n=46). Myocardial TBARS, an index of myocardial
oxidative stress, was increased in the WT+MI group,
to about 70 nmol/g, but this increase was inhibited
in the TG+MI group (40 nmol/g). Survival was improved
in the TG+MI group compared to the WT+MI group (about
85% versus 60%, p<0.01). Infarct size measured
by TTC staining showed no significant different between
the groups for the risk area (about 40% in each) or
the infarct area (about 80% in each).
|
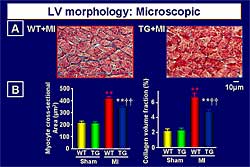 |
| Figure
4. Morphology of the left ventricle in the wild-type
and transgenic mice post-myocardial infarction.
|
| Click
to enlarge |
|
Morphology
obtained from the hearts 4 weeks after coronary arterial
ligation showed that myocyte hypertrophy and interstitial
fibrosis were increased in the WT+MI group, but were
inhibited in the TG+MI group (Figure
4). Apoptosis was inhibited by GSHPx. The increase
in TUNEL-positive myocytes in the remote area and borderline
area seen in the WT+MI mice (to about 90%) was inhibited
in the TG+MI group (about 55%). DNA ladder formation
detected by PCR was inhibited in the TG+MI group but
not in the WT+MI group. Matrix metalloproteinase-9 (MMP-9)
is involved in the pathophysiology of left ventricular
remodeling. The increased MMP-9 zymographic levels seen
in the WT+MI group was inhibited in the TG+MI group.
|
|
Study with MtTFA in transgenic mice
In the transgenic mice, human mtTFA was highly expressed
in the heart, assessed by Western blot analysis. Importantly,
endogenous mouse mtTFA was not altered in transgenic
mice. After MI was created in the mtTFA transgenic
mice and WT mice, none of the transgenic mice died,
despite comparable MI size to the WT mice.
|
|
Conclusion
Current work by this group includes the physiology,
pathology, and molecular biology in these transgenic
mice. The preliminary results are very promising,
showing that mtTFA is a very novel and promising target
in the treatment of heart failure. Based on the current
results, mtTFA can be a novel target to maintain mitochondrial
biogenesis and function and ultimately for the treatment
of heart failure.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2004
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|