 |
|
|
|
| Emerging Issue in Cardiology for
Asia |
|
|
|
|
|
Emerging
Issues in Cardiology for Asia: An Overview
Masayasu Hiraoka
Tokyo Medical and Dental University,
Tokyo, Japan
|
|
Many genetic causes of ion channel diseases have been revealed
in recent years. These diseases include the long QT syndrome
(LQTS), caused by genetic defects in potassium and sodium
channel genes, and the Brugada syndrome, which has a higher
prevalence in Southeast Asia. A sodium channel mutation
(SCN5A) causes progressive cardiac conduction disturbance
(PCCD). Catecholaminergic polymorphic ventricular tachycardia
(CPVT) is caused by 2 types of mutations, a ryanodine receptor
(RyR) gene defect and a CASQ2 gene defect. A short QT syndrome
(SQTS) has been identified recently, which might be caused
by a mutation in the calcium channel (KCNQ1), but further
study is necessary. Familiar atrial fibrillation due to
a KCNQ1 potassium channel has been identified in a large
family in China.
|
|
Brugada syndrome
First reported by Brugada and Brugada in 1992, the Brugada
Syndrome is characterized by right bundle branch block (RBBB),
persistent ST elevation in the V1-V3 leads, and sudden cardiac
death (SCD). Ventricular fibrillation (VF) occurs in patients
without organic heart disease, and there are findings of
conduction disturbances, for example, H-V prolongation.
VF attacks are prone to occur at night or during sleep,
and are unrelated to exercise. Occasionally there is a family
history of SCD.
In Southeast Asian countries, there is a relatively high
prevalence of Sudden Unexplained Nocturnal Death Syndrome
(SUNDS), characterized by SCD at night or during sleep.
In 1997, Nademanee and colleagues reported the high incidence
of SUNDS (called Lai-tai) in Thailand. Pokkuri disease has
been reported in Japan and Bangungut in the Philippines.
The clinical characteristics of Pokkuri disease include
young and middle-aged men without apparent disease who die
suddenly, mostly while sleeping at night, and without any
known factors precipitating cardiac arrest. On autopsy,
there is a patent coronary artery and no gross anomaly.
In some cases, scattered fibrosis in the conduction system
and abnormal sinus node artery is found.
A case was reported in 1976 in the Japanese Circulation
Journal of a 27-year-old man who died suddenly while hospitalized
for orthopedic surgery. His pre-operative ECG revealed a
RBBB and ST elevation in the V2 lead, identifying Brugada
syndrome in this patient.
Aihara and colleagues from the National Cardiovascular
Center in Japan in 1990 reported 4 cases of idiopathic VF.
Brugada-type ECG changes were found in 3 of the cases. A
survey of 62 Japanese hospitals in 2000 revealed 216 cases
of Brugada syndrome and 357 cases suspected to be Brugada
syndrome, and 126 cases of idiopathic VF without Brugada-type
ECG changes. Tohyo and colleagues reported in 1995 that
1 of 2000 healthy persons has Brugada-type ECG changes,
and in 2001 Atarashi and colleagues reported that the clinical
course is generally benign in asymptomatic cases with Brugada-type
ECG changes.
The ECG characteristics of the Brugada syndrome include
ST elevation in leads V1-V3, fluctuation of ST elevation
(coved type, saddleback type, no elevation), with various
factors affecting ST elevation. Generally, but not always,
RBBB is present, and some patients do not have an S wave
in the left precordial leads (pseudo RBBB or J-waves). Frequently,
Brugada syndrome is associated with left axis deviations.
|
|
Sodium channel hypothesis
Disturbances in the sodium or calcium channel may be a
mechanism for the ST elevations seen in Brugada syndrome,
as shown by experiments with class Ic antiarrhythmic agents
which are effective in ischemia.
Phase II reentry produces polymorphic ventricular tachycardia
(PVT). Phase II reentry is caused by a large difference
in voltage that can produce an extra action potential. The
difference in voltage is a result of the a result of the
prominent notch and dome-type action potential that can
occur between action potentials, as shown by an experiment
of of simulated ischemia that revealed a prominent notch
in 4 different action potentials, and that some portion
of the action potential is aborted when simulated ischemic
solutions are given.
This experimental data also explain the Brugada-type ST
elevations. In the normal setting, in the right ventricular
outflow tract, the action potential in the epicardium displays
a notch just after the peak of the action potential and
is repolarized earlier than the endocardia action potential
or M cell action potential. Also, this endocardia action
potential does not have a prominent notch, because epicardium
has abundant Ito and endocardia has the least Ito development.
In contrast, in the setting of Brugada syndrome with saddleback-type
ST elevation, when giving a sodium or calcium channel blocker,
the action potential peak is decreased in the epicardial
action potential and there is a large notch and a plateau.
Because of this large notch, the endocardial action potential
changes little, resulting in some difference in voltage
and causing the J-wave type of ST elevation. In the coved-type
ST elevation in Brugada syndrome, a further decrease of
the sodium channels, may result in a much larger notch and
delayed repolarization and negative T waves. In some areas
of the epicardium, there is a voltage action potential and
on the other side a dome remains, and this difference results
in Phase II reentry and causes the development of PVT.
In support of this sodium channel hypothesis, Chen and
colleagues reported the genetic basis and molecular mechanism
for idiopathic VF in Nature in 1998. They showed that Brugada
syndrome had autosomal dominant inheritance, a mutation
in SCN5A in some patients, and that some of these mutant
channels do not express channel function or decreased function
when expressed in some cell lines.
Mutations in sodium channel genes cause Brugada syndrome
in at least some patients, and also causes LQTS3 and progressive
conduction disturbances. This is now called sodium channelopathy
involving the SCN5A gene and is a new entity causing disease,
including LQT3, Brugada syndrome, and progressive conduction
disturbances. There are some intermediate conditions. For
example, 1795insD in SCN5A produces phenotypes of LQT3 and
Brugada syndrome. Makita and colleagues and Hiraoka and
colleagues have shown that in idiopathic VF there is a mutation
in S1710L, which has electrophysiological characteristic
in between Brugada and progressive cardiac conduction disturbances.
Sodium channel mutation cause several different arrhythmogenic
diseases.
|
|
Inherited LQT Syndrome
The Romano-Ward Syndrome (RW) and the Jervelle and Lange-Nielsen
Syndrome (JLN) are the 2 types of LQTS. The former is characterized
by autosomal dominant inheritance and normal hearing and
the latter by autosomal recessive inheritance and hearing
disturbances. Seven different channel genes causing the
LQTS have been identified so far. The 3 most prominent genes
are LQT1, caused by the mutation in KvLQT1 or KCNQ1; LQT2
caused by mutation in HERG or (KCNH2); and LQT3, caused
by mutation in SCN5A. LQT1 and LQT2 comprise 80-85% of the
LQTS. The potassium channel genes have loss of function
mutations and the sodium channel genes have gain of function
mutations in this setting.
In Japan, LQT1 and LQT2 occur with similar frequency. Most
of the functional characterizations are done in HERG or
KCNH2 mutations. Some of the findings related to LQT in
Japan are novel compared to those in Caucasians and reported
in the literature. LQT1, LQTS2, and LQT3 have a different
dominant-negative suppression, and the degree of the functional
defect is different depending on the site of the mutations.
In a survey of electrophysiological characteristics of
HERG mutations in Japanese LQT2 patients, Hiraoka showed
there were differences in the mechanism or the current suppression
depending on the site of the mutations. However, in contrast
to Brugada syndrome, the incidence and prevalence do not
seem to be different in ethnic populations compared to Caucasians.
Similar to reports from other countries, LQT1 and LQT2 have
a nearly equal incidence in Japan and LQT3 is rather rare,
and the other types of LQTS are even more rare. The types
and locations of gene mutations are somewhat different among
different races. But, there are certain genotype and phenotype
correlations among these LQTS patients.
|
PAGE
TOP
|
Genotype-
and Mutation Site-Specific Differences in Arrhythmic
Risk and Sensitivity to Sympathetic Stimulation in
the Long QT Syndrome
Wataru Shimizu
National Cardiovascular Center,
Suita, Japan
|
|
Congenital Long-QT (LQT) syndrome is a hereditary
disorder characterized by a prolonged QT interval
and polymorphic ventricular tachycardia (pVT) known
as Torsade de Pointes, mainly as a result of increased
sympathetic tone during exercise or mental stress.
Genetic studies have identified 7 forms of congenital
LQT syndrome, caused by mutations in potassium or
sodium channel genes or in membrane adaptor located
in chromosome 3, 4, 7, 11, 17, and 21. LQT-1, LQT-2,
and LQT-3 syndromes comprise more than two-thirds
of the genotyped patients. Therefore, genotype-phenotype
coordination in the LQT-1, 2, and 3 syndromes are
more clinically important for effective management
and treatment of genotyped patients.
An international registry conducted by Schwartz and
coworkers demonstrated differential triggers for cardiac
events between the LQT 1, 2, and 3 syndromes. Exercise-related
events seem to dominate the clinical picture in LQT1.
Swimming is a specific trigger in LQT1. A sudden startle
in the form of an auditory stimulus is a predominant
trigger in LQT2, and more recently LQT2 females were
shown to have a greater risk of cardiac events during
the post-partum period than the other forms of LQT
syndrome. In contrast, sleep-related events are seen
more often in LQT3. Differential sensitivity of each
genotype to sympathetic stimulation is considered
the reason for the differential triggers for cardiac
events.
|
|
Epinephrine for provocative testing
Provocative testing using epinephrine infusion or
isoproterenol infusion has been used to unmask concealed
forms of congenital LQT syndrome. In a study conducted
by this group, a bolus injection of epinephrine (0.1
mcg/kg) was immediately followed by continuous infusion
(0.1 mcg/kg/min). The 12-lead ECG was continuously
recorded during sinus rhythm (SR) and the corrected
QT-interval (QTc) was measured under baseline conditions.
The peak epinephrine effect is seen usually 1-2 minutes
after initiating the epinephrine when the heart rate
is maximally increased, and a steady-state epinephrine
effect is seen usually 3-5 minutes after initiating
epinephrine.
In LQT1 patients, in their study, epinephrine dramatically
prolonged the QTc interval at the peak epinephrine
effect. Notably, the QTc remained prolonged during
the steady-state epinephrine. The paradoxical prolongation
of the QT interval was seen in this setting. In LQT2,
the QTc interval was also prominently prolonged at
the peak epinephrine effect but returned close to
the baseline level during steady-state epinephrine.
In LQT3, the QTc interval at the peak epinephrine
effect was much less than that seen in LQT1 or 2,
and the QTc was shortened to below the baseline level
during steady-state epinephrine.
|
|
Prospective study of value of epinephrine
testing
To test their hypothesis that epinephrine
testing is useful for improving clinical ECG diagnosis
and for predicting genotypes in the LQT1, 2, and 3
syndromes, they conducted a prospective study. In
the study, there were 31 LQT1 patients, 23 LQT2 patients,
6 LQT3 patients, and 30 control patients. No significant
differences were seen between the 4 groups for the
clinical characteristics, except for the baseline
QTc interval, which was significantly longer in the
LQT2 and LQT3 patients than that in the LQT1 patients,
but all were significantly longer than that in the
control group.
|
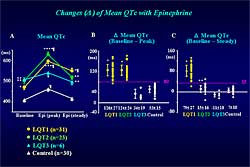 |
[Heart
Rhythm 2004;276-284]
Figure 2. The changes in mean QTc with epinephrine. |
| Click
to enlarge |
|
The study results
are presented in Figure
2. Panel A reports composite data in the change
in the QTc interval under the baseline condition at
the peak epinephrine effect and at steady state epinephrine
effect. In LQT1, the QTc was dramatically prolonged
at the peak epinephrine effect and remained prolonged
at the steady state. In LQT2, the QTc was prominently
prolonged at the peak epinephrine effect but returned
close to the baseline levels at the steady state. In
LQT3, the prolongation at peak was much less in LQT3
and control patients, and the QTc was shortened to the
baseline levels. |
|
Panel B illustrates the change of the mean QTC between
baseline and the peak epinephrine effect. The delta
mean QTC at peak was not different between LQT1 and
LQT2, but both were significantly greater than those
in the LQT3 and control groups. The delta mean QTc
at peak of  80
ms differentiated LQT1 and LQT2 from LQT3 or control. 80
ms differentiated LQT1 and LQT2 from LQT3 or control.
Panel C illustrates the mean change in QTc between
baseline and steady-state epinephrine effect. The
delta mean QTc at steady state was significantly greater
only in LQT1 than in the other 3 groups. The delta
mean QTc at steady state of 35
ms differentiated LQT1 from the other 3 groups.
The sensitivity for identifying LQT-1 mutation carriers
among the LQT1 and the control groups was relatively
low at 68% by ECG diagnostic criteria or for an LQT
score 4.
The specificity was always 100%. Thus, about one-third
of LQT-1 mutation carriers will be missed by the ECG
diagnostic criteria at baseline. The sensitivity was
substantially improved with the steady-state epinephrine
effect. The sensitivity was 87% by ECG criteria and
81% for an LQT score 4.
In contrast, for the LQT2 and LQT3 groups, the sensitivity
was relatively high, more than 80%, by ECG diagnostic
criteria. The sensitivity was further increased by
epinephrine steady-state in LQT2, however, the sensitivity
was not changed with epinephrine in the LQT3 group.
|
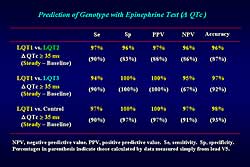 |
[Heart
Rhythm 2004;1:276-284]
Figure 3. The prediction of LQT genotype with
epinephrine testing. |
| Click
to enlarge |
|
Figure
3 illustrates the specificity, sensitivity, positive
predictive value, and negative predictive value, and
predictive accuracy based on delta QTc for genotype
prediction. The delta mean QTc at steady-state epinephrine
35
ms differentiated LQT1 from the LQT2, LQT3 or control
groups, with predictive values of more than 90%. Even
when calculating the predictive values by delta QTc
simply measured from the ECG lead V5, the predictive
accuracy was still more than 80%. The delta mean QTc
at peak epinephrine effect of 80
ms differentiated LQT2 from the LQT3 or control groups,
with a 100% predictive value.
Thus, epinephrine infusion is a powerful test to
improve clinical diagnosis of genotype-positive patients,
especially in the LQT1 syndrome. The epinephrine test
is also effective to predict the genotype of the LQT1,
2, and 3 syndromes.
|
|
Mutation site-specific study in Japanese patients
with LQT-1 syndrome
|
|
More recently, mutation site-specific differences
in clinical phenotype have been evaluated in each
genotype. Moss and colleagues reported that LQT2 patients
with mutations located in the pore region had a greater
risk of cardiac events than those with none-pore lesion
mutations including C-terminus or N-terminus. This
group examined the arrhythmic risk and sensitivity
to sympathetic stimulation between a Japanese LQT1
population with transmembrane mutations and a population
with C-terminal mutations. The study population comprised
95 LQT1 patients from 37 unrelated families collected
from 5 Japanese institutions. Of the 95 study patients,
66 patients from 27 families had a total of 19 transmembrane
mutations, and 29 patients from 10 families had 8
C-terminal mutations.
|
 |
[J
Am Coll Cardiol 2004;44:117-125]
Figure 4. Methods (details of assessments made
in mutation site-specific study) |
| Click
to enlarge |
|
Comparisons were made for clinical ECG diagnosis,
baseline 12-lead ECG measurements, cardiac events,
therapy, cumulative event curves, and exercise treadmill
testing (detailed in Figure
4). The exercise treadmill testing was conducted
in 33 patients with transmembrane patients and 16
with C-terminal mutations.
Only 4 mutations were located in the pore region.
Previous reports have shown that patients with C-terminal
mutations have a milder clinical phenotype. Therefore,
they compared patients with transmembrane mutations,
including pore regions, and those with C-terminal
mutations.
|
|
There were no differences between the 2 groups for
gender, percentage of proband, and age at the ECG-recording.
Patients with transmembrane mutations were more frequently
diagnosed to have LQT syndrome. The LQT score, using
the scoring by Schwartz and colleagues, was significantly
higher in patients with transmembrane mutations. The
baseline 12-lead ECG parameters, including QT end,
QT peak, T peak to end interval reflecting transmural
dispersion, both corrected and uncorrected, were significantly
greater in patients with transmembrane mutations than
those with C-terminal mutations. The frequency of
T wave alternans was significantly higher in patients
with transmembrane mutations.
The patients with transmembrane domain mutations
versus those with C-terminal mutations had more frequent
cardiac events (55% vs 21%, p=0.002), syncope (55%
vs 21%, p=0.002), and aborted cardiac arrest or sudden
cardiac death (15% vs zero, p=0.03). Therapy was more
frequently initiated in patients with transmembrane
domain mutations versus those with C-terminal mutations,
including more beta blockers (45% vs 21%, p=0.02).
The Kaplan-Meier cumulative event curves showed a
significant difference for all patients, with a higher
risk of a first cardiac event in patients with transmembrane
domain mutations than those with C-terminal mutations
(0.65 vs 0.25, respectively, p=0.005). Most of the
cardiac events in patients with transmembrane mutations
occurred before 15 years of age, whereas the one-half
of the patients with C-terminal mutations experienced
cardiac events after 15 years of age.
Examining the ECG lead V5 before and after treadmill
exercise testing showed that the baseline corrected
QT, QT peak, and T peak to end were significantly
greater in the patients with transmembrane mutation
than in the patients with C-terminal mutation. Moreover,
the corrected T peak to end was more prominently increased
with exercise testing in patients with transmembrane
mutations.
Changes in the 12-lead ECG parameters before and
after exercise testing, showed that corrected QT end,
QT peak, and T peak to end were significantly greater
in patients with transmembrane mutations than with
C-terminal mutations. The corrected QT end and T peak
to end were significantly increased with exercise
testing in both transmembrane mutations and C-terminal
mutations. However, they were more prominently increased
with exercise testing in the patients with transmembrane
mutations.
|
|
Conclusion
Genotype prediction of the LQT1, 2, and 3 syndromes
may be possible by the differential response of the
QTc interval to epinephrine testing. The LQT1 patients
with transmembrane mutations are at higher risk of
cardiac events and have a greater sensitivity to sympathetic
stimulation that the patients with C-terminal mutations.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2004
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|