|
|
||||||
|
|||||||
|
|||||||
|
|||
|
|||
Short and Long Term Effect of the Combination Therapy of Epoprostenol and Bosentan for Patients with Pulmonary Arterial HypertensionKatsumasa MiyajiOkayama Medical Center, Okama, JapanPulmonary arterial hypertension (PAH) remains an incurable disease. Recently, however, there has been remarkable improvement in prognosis associated with epoprostenol infusion therapy. In this lecture, Dr. Katsumasa Miyaji of Okayama Medical Center focused on results from studies at his institution with the combination therapy of epoprostenol and bosentan for PAH, particularly in patients already treated with epoprostenol. A marked improvement in survival associated with epoprostenol was demonstrated in 30 patients at Okayama Medical Center, 90% survival at 3 years and 71% survival at 5 years, and about a 60% survival at 5 years in 214 patients across Japan. This is in stark contrast to the rapidly declining rates of survival with conventional therapy reported by Barst in 1994. Combination therapy with drugs with different mechanisms of action seems reasonable and effective for treatment of PAH. These include bosentan, an endothelin receptor antagonist, and sildenafil, a phosphodiesterase V inhibitor.
Study of short- and long-term efficacy of epoprostenol and bosentan Miyaji and colleagues investigated the long-term effectiveness of combination therapy with epoprostenol and bosentan. Its short term effect of tending to improve hemodynamics at 16 weeks was shown in the BREATHE-II study as reported by Humbert et al in 2004.
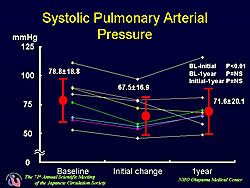
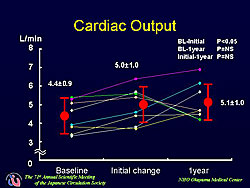
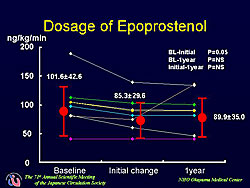
A total of 16 patients with PAH receiving intravenous epoprostenol were given oral bosentan 62.5 mg twice daily. Three patients were excluded secondary to thrombocytopenia, liver dysfunction, or aggravation of PAH and 5 patients were excluded because epoprostenol was introduced shortly after the start of bosentan therapy. In the 8 patients in the present analysis, the mean dose of epoprostenol was 102 ng/kg/min. Hemodynamics were assessed before combination therapy, a few days thereafter, and at 1 year by performing right heart catheterization. Remarkable improvement was seen in the short-term after addition of bosentan. A significant 14% decrease (from 79 to 68 mmHg) in systolic pulmonary arterial pressure was found (p<0.01). Cardiac output increased significantly from 4.4 to 5.0 L/min (p<0.01 vs baseline). However, the dose of epoprostenol had to be reduced because nearly all patients complained of symptoms related to the increase in cardiac output. The mean reduction was 15 ng/kg/min, but was 50 ng/kg/min in one patient. In the long-term, the improvement in pulmonary arterial pressure (Figure 1) and cardiac output (Figure 2) was maintained, but no further improvement was gained. The dose of epoprostenol was maintained in all but one patient in whom it was increased (Figure 3). Hence, continuous hemodynamic improvement was achieved without increasing the dose of epoprostenol and bosentan. Additional effects, such as antiproliferation or the reversal of vascular remodeling, seemed not to be obtained, even with long-term combination therapy. The improvement achieved after combination therapy is initiated with epoprostenol and bosentan can predict the long-term effect. |
Utility of Sildenafil in Pulmonary HypertensionToru SatohKeio University School of Medicine, Tokyo, JapanThe results of two studies in pulmonary arterial hypertension (PAH), one with epoprostenol and one with sildenafil, conducted by Dr. Toru Satoh and colleagues at Keio University School of Medicine were presented in this lecture, along with a comparison of their effects. This group has been using epoprostenol since 1999 and sildenafil, a PDE inhibitor, to treat PAH since 2002.
Study one: Comparison of effects on hemodynamics with sildenafil and epoprostenol Consecutive patients with idiopathic PAH (IPAH) and collagen PAH (CPH) who visited Keio University Hospital since 1999 and who were treated with epoprostenol or sildenafil monotherapy for more than 1 year underwent appropriate hemodynamic evaluation before and 1 year after treatment was initiated. This analysis comprised the patients with similar hemodynamic severity, i.e., pulmonary vascular resistance (PVR) < 20 units. Measurements included right heart catheterization with hemodynamic measurement of PVR, mean pulmonary artery pressure (meanPA), and mean right atrial pressure (mRA). Cardiac output was measured using the Fick method. Sildenafil 75 mg daily or 37.5 mg daily was given, divided into three doses depending on blood pressure. Epoprostenol was administered by an ambulatory indwelling central venous catheter, with the dose increasing over a period of 12 month to reach 40 ng/min/kg.
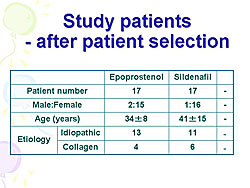
IPAH or CPH was identified in 37 of the 66 patients who received epoprostenol as a vasodilator and in 17 of the 43 patients who received sildenafil. This analysis comprises the 17 epoprostenol patients and 17 sildenafil patients whose PVR < 20 units after 1 year of treatment. The patient characteristics are detailed in Figure 1. A significant decrease in PVR after 1 year was seen with both drugs. The average decrease with epoprostenol was from about 17 units to 11 units (p<0.05), and with sildenafil from about 14 units to 11 units (p<0.05). The meanPA was significantly reduced after 1 year with both drugs, with a greater decrease with epoprostenol (from 60 mmHg to 48 mmHg; p<0.05) compared to sildenafil (52 mmHg to 46 mmHg; p<0.05). A trend towards a greater reduction in mRA was seen with sildenafil (from about 8 mmHg to 5 mmHg; p<0.06) compared to epoprostenol (about 7 mmHg, no change), although neither drug significantly decreased mRA at 1 year compared to baseline. No statistical difference in the percent change in PVR (the ratio of PVR before and after 1 year of drug treatment) was seen with either drug, although PVR tended to decrease more with epoprostenol than with sildenafil. Epoprostenol was more effective in reducing pulmonary arterial pressure after 1 year of treatment, as shown by the significantly greater decrease in the percent change in mPA with epoprostenol compared to sildenafil (p<0.05). No significant between the two drugs was seen for the percent change in mRA, although a tendency of a greater decrease with sildenafil was seen. Although epoprostenol improved pulmonary hemodynamics more, sildenafil tended to reduce right atrial pressure more, suggesting that sildenafil may have a greater effect to protect the right ventricle. In conclusion, this study showed that epoprostenol was more effective in improving hemodynamics in moderate PAH, whereas sildenafil may be more favorable for improving right heart failure. A 2005 report in Nature Medicine also suggested that sildenafil is very effective in heart failure, showing there was significantly less dilation and better wall motion in animals with pressure overload heart failure treated with sildenafil.
Study two: Effect of sildenafil in PAH unresponsive to epoprostenol Patients had PAH of any etiology or chronic thromboembolic PAH and were treated for more than 1 year with combination therapy. The 45 patients were stratified as deteriorated (n=17; mRA pressure > 8 mmHg after ≥ 6 months of epoprostenol) or improved (n=28). Hemodynamics were measured before and after epoprostenol treatment and before and after sildenafil was added to treatment. A greater decrease in the change in PVR after epoprostenol treatment compared to baseline was seen in the improved group compared to the deteriorated group. The mRA increased more than 8 mmHg after epoprostenol in the deteriorated group. Of the 17 patients in the deteriorated group, 4 patients died without other vasodilators added, 3 patients were stable without other vasodilators added, and 10 patients were given sildenafil. Five of the 10 sildenafil-added patients had improvement or stabilization of change in mRA and 5 of them deteriorated. Hence, Satoh concluded that the addition of oral sildenafil can improve patients with PAH refractory to epoprostenol. |
Nicorandil Attenuates Monocrotaline-Induced Pulmonary Arterial Hypertension in Rats: The Promising Therapeutic Potential of a Novel Combination TherapyMakato SaharaUniversity of Tokyo Graduate School of Medicine, Tokyo, JapanStudies conducted by Dr. Makato Sahara and colleagues at the University of Tokyo Graduate School of Medicine have shown that nicorandil attenuates the development of monocrotaline (MCT)-induced pulmonary arterial hypertension (PAH) in rats, via a mechanism involving the protection of pulmonary microvasculature by enhanced expression of eNOS. Further, other research by this group suggests that the combination of nicorandil and vardenafil may have a promising therapeutic potential for PAH. Nicorandil is a unique hybrid drug with nitrate-like and KATP channel activating properties. In addition to its antianginal effects, it exerts cardioprotective effects on ischemic myocardium due to its action as a KATP channel opener. However, its effect on pulmonary vasculature relating to pulmonary hypertension is not clarified.
Rat in-vivo study of nicorandil To evaluate the efficacy of nicorandil on pulmonary hemodynamics and vasculature in rats with MCT-induced PAH, Sahara and colleagues conducted an in-vivo study in rats.
Ten groups of rats were compared (Figure 1). Group 1 was normal control rats. The other nine groups, comprised of wild male Sprague-Dawley rats were injected intraperitoneally with 60 mg/kg MCT and given different treatments: 1) vehicle, 2) nicorandil 2.5 mg/kg/day, 3) nicorandil 5.0 or nicorandil plus vardenafil, 4) nicorandil 7.5 mg/kg/day, 5) nicorandil plus glibenclamide, a KATP channel blocker, 6) nicorandil plus L-Name, a NOS inhibitor, 7) vardenafil, a phosphodiesterase inhibitor, 8) nicorandil plus vardenafil, and 9) ZVAD-fmk, a caspase inhibitor that interferes with apoptosis. Vehicle, nicorandil, glibenclamide, and vardenafil were administered continuously by implanted subcutaneous osmotic pumps. At day 28, right ventricular systolic pressure (RVSP) was measured and the lungs were harvested. RVSP was significantly increased in the vehicle group. Nicorandil attenuated the progression of MCT-induced PAH in a dose-dependent manner. The beneficial effect of nicorandil was markedly inhibited by the coadministration of glibenclamide or L-NAME. Vardenafil or ZVAD also attenuated the increase of RVSP, and a combination of nicorandil and vardenafil attenuated the increase of RVSP more significantly than each drug alone. Systemic blood pressure in the vehicle group, compared to normal controls, tended to be lower (about 120 mmHg vs 138 mmHg), but was not statistically significant. Systemic hypotension was not caused by nicorandil or nicorandil plus vardenafil in rats with MCT-induced PAH. Significant medial wall thickening of pulmonary arterioles (Pas) was induced by MCT (about 60% thickening in vehicle group), and this was significantly attenuated with nicorandil 5.0 mg/kg/day (about 38% ) and nicorandil 7.5 mg/kg/day (about 35%). Glibenclamide and L-NAME markedly inhibited the effect of nicorandil. Vardenafil also attenuated the thickening of PAs (about 38%) and the attenuation with nicorandil plus vardenafil (about 30% thickening) was significantly greater than with either drug alone. In pulmonary endothelium of MCT-injured lungs, nicorandil was shown to prevent medial wall thickening, recruitment of macrophages into perivascular areas, and protected eNOS expression. Further, nicorandil upregulated the eNOS protein level and attenuated the increased expression of the activated caspase-3, which was blocked by glibenclamide or L-NAME. Notably, nicorandil plus vardenafil, compared to nicorandil alone, more significantly upregulated the eNOS protein and attenuated the expression of the activated caspase-3 in MCT-injured lungs. Furthermore, nicorandil or the combination activated the phosphorylation of Akt, a survival factor, in MCT-injured lungs.
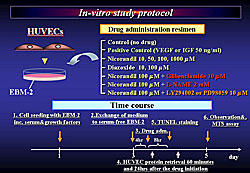
In vitro study with HUVEC
Human umbilical vein endothelial cells (HUVECs) were cultured on a 24- or 96-well plate and then incubated in serum-free medium. After 4 hours, several different drugs, including nicorandil, were administered: 1) control, 2) positive control (VEGF or IGF 50 ng/ml), 3) nicorandil 10, 50, 100, or 100 µM, 4) diazoxide 10, 100 µM, 5) nicorandil 100 µM plus glibenclamide µM, 6) nicorandil 100 µM plus L-NAME 2 mM, and 7) nicorandil 100 µM plus LY 294002 or PD 98059 100 µM (Figure 2). At 60 minutes and 24 hours post-drug administration, HUVECs protein was retrieved. HUVECs cultured in serum-free medium displayed an apoptotic morphology characterized by cell shrinkage on TUNEL staining at 8 hours and a decrease in cell viability on MTS assay. Endothelial cell death was inhibited in a dose-dependent manner with nicorandil, diazoxide, a KATP channel opener, and VEGF, the positive control. In contrast, the coadministration of glibenclamide or L-NAME reversed the effect of nicorandil. Similarly, a great number of HUVECs cultured in serum-free medium for 12 hours displayed apoptosis on TUNEL staining, which was partially attenuated by nicorandil. In contrast, the coadministration of glibenclamide reversed the effect of nicorandil.
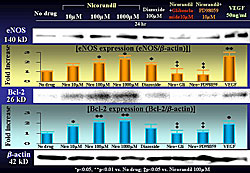
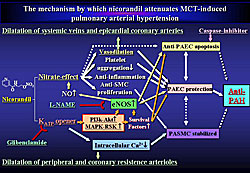
Nicorandil induced Akt and ERK phosphorylation HUVECs, which was suppressed by coadministration of glibenclamide, PI3K, or MAPKK inhibitor. Nicorandil also induced phosphorylation of an apoptosis accelerator, Bad, which is an inactive form. Bad is a downstream target of the MAPK-RSK or PI3K-Akt pathway. Further, in HUVECs treated with drugs for 24 hours, nicorandil upregulated the protein levels of eNOS and an apoptosis inhibitor, Bcl-2, a downstream target of MAPK-RSK or PI3K-Akt pathway (Figure 3). Endothelial dysfunction or damage has been hypothesized to play an important role in triggering PAH. Consequently, the mechanism by which nicorandil attenuates MCT-induced is considered to be as shown in Figure 4. In brief, nicorandil exerts a protective effect on the pulmonary endothelium via its nitrate effects, leading to the anti-PAH effects. In addition, probably via the KATP-channel opening effect, nicorandil activates the survival signaling cascades, PI3K-Akt and MAPK-RSK pathways, leading to the enhanced expression of survival and anti-apoptotic factors, and eNOS in pulmonary vasculature, which may be associated with the anti-PAH effects by nicorandil. It is known that phosphodiesterase-5 (PDE5) inhibitors exert an anti-PAH effect via the NO-cGMP pathway. A combination of nicorandil and vardenafil may have a therapeutic advantage for PAH in a synergistic manner, because the main mechanisms exerting the anti-PAH effect are considered to be different. Miyaji presented a case of a 56-year-old man with a 12-year history of primary PAH with NYHA class IV symptoms whose exercise tolerance, pulmonary hemodynamics and long-term survival significantly improved with the oral combination therapy of sildenafil, a PDE3 inhibitor, pimobendan, and nicorandil. The patient had not been responsive to calcium channel blockers or prostacyclin and was only partially responsive to a PDE5 inhibitor. |

|
|
|
Copyright © 2007 Japanese Circulation Society All Rights Reserved. webmaster@j-circ.or.jp |