Long-term lipid lowering by diet was shown to reduce
macrophage accumulation, MMP expression and activity,
and in parallel increase interstitial collagen content
in rabbit atheroma.
After 4 months of the atherogenic diet, rabbit
aorta contained a large number of macrophages, but
after 16 months of lipid lowering macrophages were
nearly undetectable. In the baseline lesion, macrophages
expressed high levels of MMP-1 or collagenase-1.
In such lesions, collagen accumulation was seen
at very low levels. However, lipid lowering reduced
collagenase expression and in parallel increased
collagen accumulation, a key determinant of plaque
stability. The conversion from a soft plaque to
a fibrous plaque was also confirmed by magnetic
resonance imaging (MRI).
Smooth muscle cell activation
Several years ago, Aikawa isolated the cDNA clones
encoding human smooth muscle myosin heavy chain
isoforms and characterized the expression pattern.
He also demonstrated that intimal smooth muscle
cells in atheroma have an immature phenotype determined
by decreased expression of smooth muscle myosin
heavy chain isoforms.
Recently, they found that in rabbit atheroma lipid
lowering promotes a more mature phenotype of smooth
muscle cells as gauged by increased expression of
myosin isoform and decreased expression of MMP.
After 4 months of hypercholesterolemia SMC in the
fibrous cap (detected by alpha-actin antibody) did
not show detectable levels of SM2 expression, a
specific marker for mature SMC, whereas medial SMC
expressed both alpha-actin and SM2. This suggests
that SMC in the fibrous cap have an immature phenotype.
However, lipid lowering by diet promoted a more
mature SMC in the fibrous cap as determined by increased
expression of SM2.
Pathologic significance of intimal SMC maturation
After lipid lowering therapy, more mature SMC expressing
SM2 showed less expression of MMP-3 and MMP-9, compared
to the baseline lesion. These results suggest that
lipid lowering can stabilize the plaque by not only
by reducing the number of macrophages but also by
promoting a more mature SMC in the intima. One candidate
mechanism for the maturation of SMC in the intima
is decreased expression of PDGF-ß
associated with a reduced number of macrophages,
since PDGF-ß is
known to suppress SMC differentiation.
Reduced prothrombotic potential
Recently this group showed that dietary lipid
lowering reduces tissue factor expression and activity
by macrophages and SMC in rabbit atheroma. Tissue
factor is a potent contributor to acute coronary
events. After 4 months of the high cholesterol diet,
lesional macrophages expressed tissue factor in
the intima. However, after lipid lowering therapy
tissue factor was nearly undetectable, associated
with a reduced number of macrophages. These results
suggest that lipid lowering can stabilize the plaque
by decreasing proteolytic and prothrombotic macrophages.
However, the mechanism by which lipid lowering decreases
macrophages remains unclear.
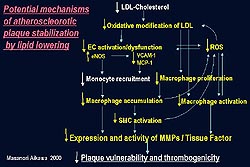
Atherosclerotic lesions produce excess reactive
oxygen species (ROS) including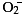 .
is known to induce oxidative modification of LDL.
A number of in vitro experiments have suggested
such oxidative stress can promote endothelial cell
activation or dysfunction and induce expression
of cell adhesion molecules such as VCAM-1 and chemokines
including MCP-1, leading to increased monocyte recruitment.
.
is known to induce oxidative modification of LDL.
A number of in vitro experiments have suggested
such oxidative stress can promote endothelial cell
activation or dysfunction and induce expression
of cell adhesion molecules such as VCAM-1 and chemokines
including MCP-1, leading to increased monocyte recruitment.
To understand the mechanisms of reduced number
of macrophages in the lesion they first measured
the level of ROS production using lucigenin chemiluminescence
assay. Aortic rings from hypercholesterolemic
rabbits elaborated high levels of ROS production,
compared to normal aorta. However, lipid lowering
decreased the production of ROS to levels similar
to those in normal aorta.
Endothelial cell activation
Further studies in the rabbit model suggest that
lipid lowering can limit endothelial cell activation
and dysfunction based on the decreased expression
of VCAM-1 and MCP-1 and increased expression of
eNOS.
Oxidized LDL is known to induce VCAM-1 expression
by endothelial cells in vitro. After 4 months of
hypercholesterolemia oxidized LDL accumulated in
the intima underlying endothelial cells overexpressing
VCAM-1. However, after lipid lowering therapy oxidized
LDL accumulation and VCAM-1 expression were nearly
undetectable.
MCP-1 is a potent monocyte chemoattractant that
can induce monocyte recruitment into the intima.
After 4 months of hypercholesterolemia MCP-1 was
detected in endothelial cells as well as SMC and
macrophages. However, after lipid lowering therapy
nearly no MCP-1 could be detected in the rabbit
lesion.
Several in vitro studies suggested that oxidative
stress can decrease the expression of endothelial
nitric oxide synthase (eNOS) by endothelial cells.
Decreased eNOS should promote macrophage-rich atheroma
formation, since eNOS can limit the monocyte-endothelial
cell interaction (Fig. 2). In rabbit atheroma after
4 months of hypercholesterolemia few if any endothelial
cells stained positively for eNOS antibody. However,
after 16 months of lipid lowering therapy eNOS expression
substantially increased.