 |
|
|
|
IS106
Genetic Manipulation of Beta-adrenergic
Signaling in the Failing Heart |
|
Walter J. Koch, Ph.D.
Department of Surgery
Duke University Medical Center
Durham, NC, USA |
|
|
|
|
|
|
|
|
The overexpression of the beta-2 adrenergic
receptor (ß-2 AR) has therapeutic potential in patients
with heart failure. Importantly, using gene therapy, the
levels that lead to pathology in transgenic mice will
not be reached. This hypothesis is supported by these
findings reviewed in this lecture.
- ß-2 AR have very specific signaling differences
from the beta-1 adrenergic receptors (ß-1 AR).
They showed in mice that ß-1 AR overexpressed
only at 10-fold led to dilated cardiomyopathy (DCM)
and pathology at 3-4 months of age.
- In the Gq mouse model, hypertrophy can be reversed
and function improved with Gaq
overexpression.
- Adenoviral-mediated ß-1 AR overexpression
in cardiomyocytes leads to apoptosis, while adenoviral-mediated
ß-2 AR overexpression does not. The present
data says that the ß-2 AR could actually protect
against ß-1 AR-induced apoptosis.
- Adenoviral-mediated ß-2 AR overexpression
in failing cardiomyocytes rescues the signaling abnormalities.
The ß-2 AR given to normal rabbit hearts in
vivo improves systolic and diastolic function.
|
PAGE
TOP
The ß-adrenergic
receptor system |
|
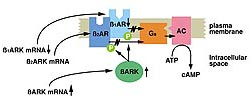 |
Figure
1. The ß-adrenergic receptor (AR) system
in heart failure. In end stage human heart failure,
the ß-1 AR mRNA and protein are selectively
downregulated, while the ß-2 AR remain the
same. The ß-1 and ß-2 receptors are
uncoupled, mediated by the beta receptor kinase
ßAR kinase (ßARK), a member of the
family of G protein coupled receptor kinases.
ßARK mRNA protein inactivity is increased
about 3-fold in human heart failure, which can
lead to the enhanced phosphorylation of ß
receptors and the functional uncoupling seen in
cardiac disease. (Circulation 1993;87:454-463.)
Click to
enlarge
|
|
The ß-adrenergic receptor system
plays a key role in cardiac function in normal and
diseased states. In end stage human heart failure,
the ß-1 AR mRNA and protein are selectively
downregulated, while the ß-2 AR are not changed
for the most part (Fig. 1). Both the ß-1 and
ß-2 receptors are uncoupled, mediated by the
beta receptor kinase ßARK, a member of the
family of G protein coupled receptor kinases. ßARK
mRNA protein inactivity is increased about 3-fold
in human heart failure, which can lead to the enhanced
phosphorylation of ß receptors and the functional
uncoupling seen in cardiac disease.
ß-2 AR activation causes coupling
to the heterochimeric G protein. Gas
can activate cyclase. The beta-gamma dimer effector
molecules can lead to activation of downstream target
events 1) ion channel activation, 2) activation
of certain forms of adenyl cyclase, 3) importantly,
specifically binding residues on the C terminus
of ßARK (ß ARKct), bringing ßARK
into more contact with the membrane where it can
phosphorylate agonist-occupied receptors. So, peptides
from this region of ßARK have been shown to
be in vivo and in vitro ßARK inhibitors.
|
|
PAGE
TOP
|
Mouse models developed in Koch's laboratory
showed that inhibiting the activity or lowering
the expression of ßARK allows direct regulation
of cardiac contractility. Two transgenic mice models
of hypercontractile function were generated using
the alpha myosin heavy chain promoter: selective
cardiac overexpression of the human ß-2 AR
and inhibition of ßARK using the inhibitor
ßARKct. Overexpression of ß-2 AR in
a dose-dependent manner led to enhanced cardiac
function and to DCM. At more modest levels of overexpression
no pathology was seen.
Their work is the first in vivo demonstration
that ßARK could be present and desensitize
beta receptors. The ßARKct peptide inhibitor
had enhanced basal dP/dtmax and supersensitivity
to isoproterenol. A reciprocal regulation of cardiac
contractility was found when they overexpressed
the ßARK-1 enzyme or the ßARKct peptide
inhibitor. ß ARK overexpressing mice had attenuated
function. Heterozygote ßARK knockout animals
with 50% less ßARK had increased basal dP/dtmax
and supersensitivity to isoproterenol, much like
the ßARKct animals.
|
|
PAGE
TOP
Studies
of ß-2 AR overexpression |
|
To determine whether inhibiting ßARK
or overexpressing receptors had any therapeutic
value, the adenoviral approach in larger animals
was used in Koch's laboratory. No mouse models of
heart failure were available at that time. The development
of DCM was prevented by the expression of the ßARKct
peptide when the MLP knockout mouse (with DCM) and
the ß ARKct animals were bred. The catalytic
domain of the ßARK-1 protein is very similar
to protein kinase A (PKA) and C (PKC). The N terminal
domain has some interesting functions that Koch's
laboratory is now delineating. The beta-gamma binding
domain is in the carboxy terminus, the region used
for the ßARKct.
Prior work has shown that the
wild type mice have normal systolic and diastolic
function on echo, whereas the MLP knockout mice
have extreme dilatation, thin walls, and very limited
fractional shortening. In the MLP-ßARKct hybrid
animal, systolic and diastolic function is normalized,
with no dilatation.
Interestingly, work by other investigators
has shown positive effects with ß-2 AR overexpression
and variable effects with ßARKct. In the Gaq
transgenic mouse in Dorn's laboratory, modest ß-2
adrenergic overexpression led to some functional
and hypertrophic rescue. In Leiden's model of idiopathic
DCM dominant negative Kreb mice, the ßARKct
had some positive inotropic effects that did not
seem to rescue the mortality in these animals; a
variable effect. Leinwand's mutant myosin heavy
chain animal with a cardiomyopathic phenotype was
rescued by ßARKct.
|
|
PAGE
TOP
CSQ overexpression
experiments |
|
Targeted CSQ overexpression led to
altered SR function and calcium handling in the
CSQ overexpressing model generated by Jones. These
mice have early hypertrophy followed by progressive
DCM. The CSQ transgenic mice at 7 weeks have concentric
hypertrophy, with normalized function that rapidly
deteriorates to a state of dilatation and low systolic
function at 14 weeks. All the mice are dead by 16
weeks; severe early mortality in the CSQ transgenic
mice in contrast to MLP knockout mice. At 7 and
14 weeks of age there was no beta receptor responsiveness
in relation to normalized function as seen by echo.
Biochemically, decreases in beta receptor density
and a 2-fold increase in ßARK protein and
activity was seen. These beta receptor abnormalities
were present at 7 weeks, before the onset of dilatation.
The CSQ transgenic have large left
ventricles as shown by the LV end diastolic dimension
and in the CSQ-ßARKct animals there was substantial
rescue. The fractional shortening that was significantly
impaired in the CSQ mice, was significantly elevated
in the hybrid animals from about 15% to about 40%
in experiments in Koch's laboratory.
Importantly, a survival study
showed that ßARKct chronically expressed in
the heart failure model lead to positive effects.
The CSQ animals die very early at 15-16 weeks of
age. In the ßARKct animals a statistically
significant increase in survival to more than 20
weeks was found. However, once the animals begin
to die the curves are nearly parallel, showing that
despite the substantial survival effect, there is
a threshold the ß ARKct can not overcome.
|
|
PAGE
TOP
|
| |
Koch's laboratory attempted to deliver
the ß-2 receptor and ßARKct to the hearts
of rabbits by intracoronary delivery. The objective
was to inhibit ßARK-1 mediated desensitization
by expressing the ßARKct to enhance cardiac
contractility and restore normal beta receptor responsiveness,
a limitation of the failing heart.
In a study using a percutaneous approach,
a subselective catheterization was done of a rabbit
coronary artery and 5 x 1011 total viral
particles of an adenovirus containing either the
ß-2 receptor, ßARKct or beta-Gal injected.
Ventricular-targeted expression of the transgene
is possible as shown by the beta gal; going down
the right coronary artery only transduces the right
ventricle, the left circumflex is distributed through
the left ventricular free wall.
|
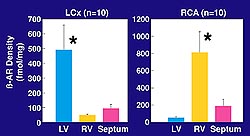 |
Figure
2. ßAR density after subselective intracoronary
delivery of Ad5/ß2 in the rabbit model.
Overexpression is seen in the left ventricle and
no expression in the right ventricle using left
circumflex delivery. Higher levels of expression
are obtained in the right ventricle using right
coronary artery delivery. ß receptor overexpression
in the range of 6- to 10-fold led to enhanced
contractility of the rabbit heart at 7-21 days
after gene delivery. (Circulation 2000;101:408-414.)
Click to
enlarge
|
|
Left ventricular overexpression, with
no overexpression in the right ventricle, was obtained
injecting the ß-2 receptor through the left
circumflex artery (Fig. 2). Higher levels of expression
are obtained in the right ventricle as it is easier
to transduce, probably due to it being thinner with
less pressure. ß receptor overexpression in
the range of 6- to 10-fold led to enhanced contractility
of the rabbit heart at 7-21 days after gene delivery.
ßARKct was also deliverable using this method.
Heart failure study
Gene delivery of adenoviral ßARKct
in a rabbit model of heart failure resulted in a
significant increase in regional fractional shortening
in the ßARKct animals, and 2-D echo showed
a slight but significant increase in fractional
shortening. Each animal served as its own control,
and serial echos showed an increase in fractional
shortening. The failing control animals that received
beta-Gal worsened or had no change.
In this study, heart failure was induced
by performing a left circumflex ligation in a marginal
branch to produce a 30-40% infarct in the LV free
wall. Hemodynamic failure developed within 3 to
4 weeks, and the ischemic model was allowed to progress
to heart failure. Percutaneous left circumflex-mediated
delivery of the adenoviral ßARKct as the ßARKct
inhibitor was performed. At one and two weeks after
gene delivery, in vivo assessment was performed
regionally using sonomicrometry crystals placed
on the plane of the LV and by 2-D and milar micromanameter
catheterization.
|
|
PAGE
TOP
ßARK-1
inhibition as a novel heart failure target |
|
Expression of ßARK in human
heart failure is associated with severity. Transgenic
mice with cardiac-targeted ßARKct expression
have enhanced cardiac performance without pathology,
and do not have a higher susceptibility to ischemic
injury. The heterozygote ßARK knockout mice
have increased cardiac function.
Heart failure has been prevented
or attenuated by cardiac expression of the ßARKct
in several mouse models of cardiomyopathy, and it
increased survival in the CSQ model. In the rabbit
model, adenoviral-mediated cardiac delivery of ßARKct
increased systolic and diastolic function in normal
hearts. Heart failure was prevented after myocardial
infarction by ßARKct delivery, as shown in
a study to be published soon. Cardiac function following
heart failure development can be improved.
The ßARKct acts as an inhibitor
of beta gamma activation of ßARK. It is known
from previous studies that ß ARKct inhibits
ßARK in the heart, beta receptor desensitization
is attenuated, and beta receptor signaling is enhanced.
However, since ßARK probably phosphorylates
hundreds of receptors, including several in the
heart, there could be other signaling systems responsible
for the phenotype
Novel paradigm of receptor signaling
Koch's laboratory is now testing the
interesting speculation that ßARK translocation
is the mechanism triggering beta gamma-dependent
MAP kinase activation. If ßARK translocation
is the contributing mechanism, it is due to ßARK
inhibition. Other labs have shown that this beta-gamma
G protein coupled receptor activation of the RAS-MAP
kinase pathway, progresses from the internalization
of receptors by beta-Arrestin, which binds GRK phosphorylated
receptors. This can not occur unless the receptor
is phosphorylated by GRK or ß ARK. The ßARKct
is likely inhibiting desensitization of the beta
receptor in the heart. Classical signaling through
cAMP through the calcium channels is enhanced, which
could support the decreased contractility of the
heart. Since it is decreasing desensitization, fewer
internalized receptors to activate MAP kinase are
available. In this novel paradigm of receptor signaling,
the desensitized receptor, classically shown to
be turned off, is actually the signaling molecule.
|
|
PAGE
TOP
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2000
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|