 |
|
|
|
IS097
Keynote Lecture
Signaling Events in Cardiomyocyte Dropout |
|
Gerald W. Dorn II, M.D.
Division of Cardiology
University of Cincinnati
Cincinnati, OH, USA |
|
|
|
|
|
|
|
|
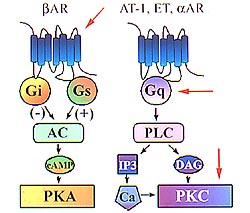 |
| Figure 1.
The beta-adrenergic signaling system. The beta-adrenergic
receptor (ßAR) may couple either through Gi or
Gs to adenylcyclase in an inhibitory or stimulatory
fashion, resulting in an increased formation of cyclic
AMP and activation of protein kinase A (PKA). In the
Gq phospholipase C pathway (PLC), angiotensin, endothelin,
alpha adrenergic receptors, prostaglandin F2-alpha,
and other agonists, each through their own independent
receptors, can activate protein kinase C (PKC). (Dorn
2000) |
|
In cardiac hypertrophy, particularly concentric
cardiac hypertrophy caused by pressure overload and in
a number of genetic models of cardiac hypertrophy, there
is increased cardiac myocyte size, giving rise to increased
ventricular mass. However, in the progression from hypertrophy
to heart failure, the seminal cellular finding is a loss
of cardiac mycocytes. The focus of this lecture was the
mechanisms whereby cardiac myocyte loss may result in
dilated cardiomyopathy (DCM). The potential for modifying
hypertrophy in heart failure by adjusting the viability
and the health of cardiac myocytes by differential regulation
of specific signaling molecules was shown in research
conducted in Dorn's laboratory.
Cardiomyocyte necrosis, cardiomyocyte apoptosis,
and cardiomyocyte hyperplasia are three cellular mechanisms
for cardiac dilation and failure. In necrosis, the external
milieu of the cardiomyocyte is so toxic it dies, leaving
remnants that result in an inflammatory response and ultimately
cardiac fibrosis. In apoptosis, the external milieu is
such that the cardiomyocyte perceives it can no longer
survive and initiates an internal program of cell death
causing the cell to degrade its own proteins and DNA.
The cell is completely removed by autolysis, with little
inflammatory response. Hyperplasia is a rather novel mechanism,
whereby insufficient numbers of cardiac myocytes are generated
during pre-natal or post-natal development and results
in dilated phenotypes.
In the beta-adrenergic signaling system
(Fig. 1), the beta-adrenergic receptor (ßAR) may
couple either through Gi or Gs to adenylcyclase in an
inhibitory or stimulatory fashion, resulting in an increased
formation of cyclic AMP and activation of protein kinase
A (PKA). In the Gq phospholipase C pathway (PLC), angiotensin,
endothelin, alpha adrenergic receptors, prostaglandin
F2-alpha, and other agonists, each through their own independent
receptors, can activate protein kinase C (PKC).Genetic
modification of Gq, PKC, and beta-adrenergic receptors
was discussed.
|
PAGE
TOP
|
| |
In five viable lines with levels
from 60- to 350-fold normal in a model of ß-2 AR
overexpression, at lower levels of ßAR overexpression
the hearts were virtually normal in pathology, normal
size, wall thickness and function; similar to initial
reports from other investigators of ß-2AR overexpression
enhancing cardiac function. Toxic levels, 350-fold
baseline, resulted in a phenotype with left and
right ventricular dilation, wall thinning, massive
atrial enlargement with formation of thrombus in
the atria-indicative of a murine DCM.
|
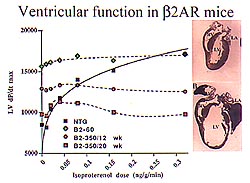 |
Figure
2. In vivo hemodynamic analysis measuring left
ventricular (LV) dv/dt as a function of isoproterenol
infused showed a parallel increase in isoproterenol
dose and LV contractile function. Basal LV function
was maximal in the lower level overexpressers.
In the higher level overexpressers, at 12 weeks
basal function was enhanced over non-transgenic
with no response to isoproterenol and function
was diminished with high levels of isoproterenol.
At 20 weeks, the functional decline was even more
enhanced. Basal function was essentially normal,
with no response to isoproterenol. (Dorn 2000)
Click to
enlarge |
 |
|
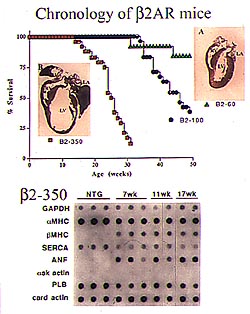
Figure
3. The survival curve from the longitudinal
analysis showed that at about 11 weeks, the
350-fold overexpressers began to die, and all
were dead by 35 weeks (top panel). Molecular
analysis using RNA dot blot expression of hypertrophy-associated
genes in a time course of 7, 11 and 17 week-old
ß-2AR-350 animals revealed no molecular
progression (bottom panel). The same molecular
abnormalities of increased beta myosin heavy
chain and atrial natriuretic factor expression
were present in 7-week and 17-week old animals.
(Dorn 2000)
Click
to enlarge
|
|
In vivo hemodynamic analysis measuring
left ventricular (LV) dv/dt as a function of isoproterenol
infused showed a parallel increase in isoproterenol
dose and LV contractile function (Fig. 2). In the
lower level overexpressers, basal LV function was
maximal. In the higher level overexpressers, cardiac
function was measured at two time points. At 12
weeks, basal function was enhanced over non-transgenic,
with no response to isoproterenol, and at high levels
of isoproterenol function was diminished. At 20
weeks, the functional decline was even more enhanced.
Basal function was essentially normal, with no response
to isoproterenol.
A time-dependent decline in LV function
in the ß-2AR mice at high levels of overexpression
was suggested by these data, prompting a longitudinal
analysis. The survival curve showed that at about
11 weeks, the 350-fold overexpressers began to die,
and all were dead by 35 weeks (Fig. 3,top panel).
This suggested the "classic" progression towards
DCM, if indeed a hypertrophy phenotype exists. Molecular
analysis using RNA dot blot expression of hypertrophy-associated
genes in a time course of 7, 11 and 17 week-old
ß-2AR-350 animals revealed no molecular progression
(Fig. 3, bottom panel). The same molecular abnormalities
of increased beta myosin heavy chain and atrial
natriuretic factor (ANF) expression were present
in 7-week and 17-week old animals, the age at which
a selection bias began to appear in the cardiac
phenotype due to death.
A rather pronounced cellular progression
was present. Full thickness LV histologic samples
showed: at 7 weeks a normal histology, at 11 weeks
areas of cardiomyocte dropout, at 17 weeks more
pronounced areas of cardiomyocte dropout with indication
of some fibrotic replacement, and at 25 weeks nearly
complete replacement of the ventricular myocardium
by fibrosis. This is progressive cardiomyocyte necrosis
and replacement fibrosis resulting in a DCM.
|
|
PAGE
TOP
|
| |
A model of peripartum DCM in a transgenic
mouse overexpressing the Gaq
signaling protein was studied. Gaq
is the first common signaling molecule for a variety
of hypertrophy-stimulating agonists, including angiotensin
I, endothelin and ß-adrenergic agonists. Gaq
overexpression at 4- to 5-fold endogenous levels
resulted in a hypertrophy phenotype with increased
LV wall thickness and size, a 20% increase in cardiac
mass, extremely increased levels of beta myosin
heavy chain and ANF expression, increased ß-skeletal
actin expression, and decreased sarcoplasmic reticulum
calcium ATP expression.
The model very closely reproduced
pressure overload hypertrophy. The animals had about
a 20% decrease in contractile function and enlarged
cardiac myocyte cross-sectional areas.
Animals with about 8-fold overexpression
of the Gaq signaling protein resulted from breeding
animals with about 4- to 5-fold overexpression.
A different phenotype resulted: chronic progressive
cardiomyopathy, quite large heart, left and right
ventricle dilation, quite large atria, and cardiomyocyte
dropout with a small amount of fibrosis. In the
peripartum period, a more severe form of the Gaq
CM is seen. An impregnated transgenic mouse developed
a severe rapidly progressing CM, generally within
one week of giving birth. The mortality rate was
about 50% per pregnancy event. The hearts are generally
about 300-fold larger than a normal Gaq heart, organized
and acute thrombus in the left and right atria can
be seen, indicating a chronic low cardiac output
state.
The involvement of an apoptotic process
was suggested by the nuclear morphology and in vitro
studies. On histology, a cellular transparency and
an associated nuclear abnormality in which the chromatin
began to clump in the periphery or the center, much
unlike the H&E stain of the normal heart was
seen. TUNEL assay, DNA laddering, and ultrastructural
examination revealed a relatively massive apoptotic
process, with apoptotic indices approaching 20%.
The mitochondrial abnormality in the Gq peripartum
cardiomyopathy models was striking ultrastructurally,
with a great deal of disarray and the internal tubular
structures falling apart, seemingly out of proportion
to the sarcomeric degeneration.
Transverse aortic coarctation
This represents another method
to progress the Gq heart to DCM. The animals were
aortic-banded, and followed for 3 weeks. In the
non-transgenic animals pre- and post-aortic coarctation
there was the expected development of concentric
hypertrophy, with an increase in the relative ratio
of wall thickness to ventricular radius. However,
in the Gq animals, an eccentric form of hypertrophy
progressed to a DCM. A small amount of hypertrophy
was seen, but was counterbalanced by an increase
in ventricular dimension.
The banded Gq model is associated
with cardiomyocyte apoptosis, as shown by recent
studies performed in collaboration with Ross. This
apoptotic process differs somewhat from the peripartum
model. The TUNEL staining occurs in clusters rather
than diffusely throughout the myocardium, and the
area of staining corresponds to areas of cardiomyocyte
dropout and the beginning of a fibrotic process.
Interestingly, the levels of cardiomyocyte
apoptosis seen in the Gq mice inversely correspond
with the depression of cardiac function after banding,
as assessed by echocardiographic LV shortening.
There is a shortening between apoptosis and diminished
LV function in these animals.
|
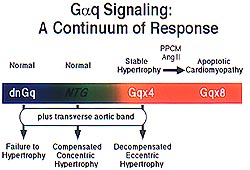 |
| Figure
4. The phenotypic progression in the Gq mice is
a continuum from normal in non-transgenic mice,
to stable hypertrophy in the Gq overexpressers,
to apoptotic cardiomyopathy in either high level
Gq overexpression, peripartum cardiomyopathy,
or after aortic banding. (Dorn 2000) |
|
In the Gq mice, there is a progression
of phenotype from normal in non-transgenic, to stable
hypertrophy in the Gq overexpressers, to apoptotic
CM in either high level Gq overexpression, peripartum
CM, or after aortic banding (Fig 4). Koch has previously
shown that dominant negative inhibition of Gq causes
failure to hypertrophy in the presence of transverse
aortic banding. Thus, it is believed that the complete
spectrum of Gq signaling and its association with
hypertrophy has been identified.
Progression of hypertrophy to apoptotic
CM
An hypothesis that a genetic program
for Gq apoptosis exists, recapitulating the genetic
program for Gq hypertrophy, was then formed. This
is rather novel, as no genetic program for apoptosis
is known. A small cluster of apoptosis genes that
is upregulated was found by comparative analysis
of the Gq and non-transgenic gene expression animals
using Incyte DNA microarrays.
The Gq animal is not undergoing apoptosis,
but is pre-disposed to apoptosis. This finding is
supported by DNA microarray analysis and conventional
assay of ANF. For the apoptosis gene caspase-1,
the relative differential expression on the chip
was 4.0 and Northern blot analysis showed substantial
upregulation. Increased levels of pro-caspase-1
can be measured in the Gq animals, relative to the
non-transgenic animals, but is not associated with
caspase activation. Upregulation of the Fas-associated
factor was found. A non-identified express sequence
tag, with substantial upregulation and some homology
to a Bcl-interacting protein, was studied. Multiple
transcripts were noted on RNA dot blot analysis.
Using that cDNA fragment, they cloned from a mouse
cardiac library mouse Nix cDNA. Nix is a recently
described pro-apoptotic mitochondrial protein, that
forms pores, interacts with Bcl-2 and Fas, and is
probably the mitochondrial effector for the pro-apoptotic
proteins. Mitochondrial degeneration is a hallmark
feature of the Gq apoptosis. Alternately spliced
variants of Nix that are differentially regulated
in the Gq mouse have been found in their lab, and
they are now looking at this gene in human hypertrophy
and heart failure. They believe they are now beginning
to understand the mechanism whereby the Gq signaling
pre-disposes to cardiomyocyte apoptosis.
|
|
PAGE
TOP
|
Through genetic manipulation they
recently have shown it is possible to modulate phenotypes
by increasing the size or number of cardiomyocytes
or by preventing their loss. Unpublished data on
epsilon PKC translocation inhibition and activation,
which can result in a hypoplastic phenotype, was
reviewed.
The Gq pathway downstream of phospholipase
C becomes quite complex due to a number of endogenous
protein kinase Cs. These may have different effects
on the cardiomyocyte, and determining which may
be effectors of the Gq or other phenotypes and which
may result in hypertrophy, heart failure or apoptosis
or a combination is difficult. Rosen has demonstrated
that the mechanism of activation of PKCs is directly
related to their location in the cell and to the
steric form of the molecule. Inactive PKC is folded
over onto itself and is associated with receptors
for inactive C kinases. When the PKCs need to be
activated by calcium or diacylglycerol (DAG), they
unfold and thereby expose their substrate binding
site and a binding site for a receptor for activated
C kinase (RACK), which can be conceived of as a
factory. The unfolded PKC goes to work in the factory
where the substrates are, allowing substrate-specific
activity of different PKC isoforms, as each PKC
isoform has a different RACK.
Rosen conceived of the notion of using
competitive peptides based on the RACK binding sequences
in active PKC to prevent their interaction with
RACK. Alternately, other peptides can be used even
in the absence of calcium and DAG to force the PKC
to unfold and expose its RACK binding site, thereby
translocating and activating it. These are catalytic-inactive
peptides that can modulate the translocation and
hence activation of PKC.
Studies with epsilon PKC
This approach has been used in Dorn's
lab to express PKC-specific peptides for epsilon,
delta, alpha, beta, and for the atypical PKCs. The
biochemical data for ePKC activating peptide, called
pseudo epsilon RACK, and an inhibiting peptide,
shows there is no effect on the overall amount of
ePKC. However, the activating peptide does increase
the amount of PKC in the particulate fraction, about
20% at baseline. The inhibitory peptide decreases
the amount of ePKC in the particulate fraction,
about 20% at baseline. The lack of an effect on
alpha PKC shows this to be an isoform-specific effect.
Epsilon PKC activation and inhibition
can increase or decrease the number of cardiac myocytes
in the heart. Activation of PKC increased the number
of cardiac myocytes that grew during the early post-embryonic
period in their studies. This activation led to
a hypertrophic phenotype with small hearts with
thick walls, functionally normal by echo, biochemical
and invasive measures. The molecular phenotype showed
an increase in beta myosin heavy chain alone; the
ANF transcript was not altered. The heart weight
to body weight was increased at 15 weeks but not
at 8 weeks in the transgenic animals. This was not
cellular hypertrophy, because the myocardial cells
were smaller than those in the non-transgenic heart.
A model of inhibition of cardiomyocyte
proliferation during post-natal development resulting
in DCM was developed through ePKC inhibition with
epsilon B-1 peptide. Lower or modest levels of expression
of epsilon B-1 peptide resulted in normal looking
hearts, whereas very high levels of expression resulted
in DCM with lethality at about 30 weeks. Histologic
examination showed cardiomyocytes to be low in number
but very large with no replacement fibrosis, as
seen in the ßAR model.
The addition of the pseudo-epsilon
RACK transgene to the Gaq transgene resulted in
a smaller heart with more cardiac myocytes, and
normal liver and lungs. Enhanced cardiac function
and increased fractional shortening in a smaller
heart with normal wall thickness resulted.
|
|
PAGE
TOP
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2000
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|