|
|
|
|
| Molecular Mechanisms of Heart
Failure |
|
|
Takashi Miyauchi, M.D.
University of Tsukuba,
Tsukuba, Japan
Yoshihiko Saito, M.D.
Kyoto University of Kyoto,
Kyoto, Japan
Masafumi Yano, M.D.
Yamaguchi University, Yamaguchi,
Japan
Seiji Takashima, M.D.
Osaka University, Osaka,
Japan
|
|
|
|
|
|
|
|
|
| New insights into the pathophysiology
of heart failure involving agonists, receptors, G proteins
and intracellular calcium mobilization were discussed in this
symposium. The insights can be integrated with genetic engineering
and conventional research methods to clarify the relation
between molecular mechanisms and the progression of heart
failure. |
Endothelin
in Heart Failure |
|
Endothelin-1 (ET-1), a potent vasocontractile peptide
produced by endothelial cells, is also produced by
cardiac myocytes. ET-1 induces cardiac hypertrophy
and cellular injury of cardiac myocytes in addition
to its potent positive inotropic and chronotropic
actions. ET-1 has arrhythmogenic actions on the heart.
It has been reported that plasma ET-1 concentrations
are elevated in heart failure in humans and in experimental
animal models. These studies suggest the involvement
of the ET-1 pathway in the pathophysiology of heart
failure.
The endothelin receptor antagonist, BQ-123, was shown
to greatly improve survival in rats with heart failure
by Miyauchi and colleagues at the University of Tsukuba.
An endothelin receptor antagonist was also shown by
this group to improve the decrease in expression of
functional molecular markers mRNA levels of the ryanodine
receptor and the sarcoplasmic reticulum CA2+-ATPase.
This suggests that one mechanism for favorable effect
of the endothelin receptor antagonist is amelioration
of the decrease in these markers and amelioration
of the calcium overload of myocardial cells.
This group has also shown that chronic treatment
with an endothelin receptor antagonist normalized
the increase in ACE mRNA in heart failure rats, and
the increase in AT1 receptor mRNA in the failing heart
of rats with heart failure due to myocardial infarction.
Therefore, suppression of the renin angiotensin system
(RAS) may be one mechanism by which the endothelin
receptor antagonist exerts its beneficial effect.
The cross-talk between the endothelin and RAS systems
was studied by this group using transgenic hypertensive
mice with hypertrophy and overexpressing angiotensin
II, which have both human renin and human angiotensinogen
due to the mating process. Because ET-I is augmented
by angiotensin II in vitro, mRNA in the hearts of
the transgenic hypertensive rats was measured. ET-1
mRNA was greatly enhanced in these mice hearts in
vivo. Chronic treatment with an endothelin receptor
blocker greatly ameliorated the hypertrophy seen in
the control mice. Therefore, in renin angiotensin
overexpressing mice, an endothelin receptor antagonist
greatly ameliorated the cardiac hypertrophy. Thus,
it can be speculated, stated Miyauchi, that angiotensin
II has a direct effect on cardiac myocytes and induces
endothelin, which causes cardiac hypertrophy.
Combined treatment with an endothelin receptor antagonist
and an ACE inhibitor improved survival to about 90%
in hamsters with heart failure, compared to about
a 45-50% survival with an endothelin receptor alone,
about a 35% survival with an ACE inhibitor, and about
a 5% survival with no treatment. This shows that both
the endothelin and RAS systems contribute independently
to the progression of heart failure and that there
is cross talk between these two systems.
Miyauchi and colleagues hypothesize that in the failing
heart, impaired myocardial energy metabolism occurs.
The impairment of beta oxidation of fatty acids causes
activation of the glycolytic system, and this switch
in metabolic energy induces increased cardiac gene
expression of ET-1. Experiments they subsequently
conducted supported this hypothesis.
An increase in oxygen consumption by cardiac myocytes
and a switch in the principle ATP energy source from
mitrochondrial fatty acid oxidation to a glycolytic
system are involved in the pathophysiology of heart
failure. Since ATP production per oxygen consumption
by glycolysis is greater than that by mitochondrial
oxidation in the failing heart, this alteration in
energy metabolism may have a compensatory aspect for
the failing heart as an adaptation. Hypoxia-inducible
factor (HIF)-1 ,
a transcriptional factor, is known to be involved
via the increased expression of glycolytic enzymes.
HIF-1
activates the gene expression of glycolytic enzymes,
which are activated as compensation for mitochondrial
oxidation of fatty acids in the failing heart. Miyauchi
and colleagues identified by sequence analysis of
the 5’-flanking promoter region of ET-1 an HIF-1
recognition site in this region. Therefore, they hypothesized
that HIF-1
is involved in the increase in the myocardial expression
of the ET-1 gene in heart failure. ,
a transcriptional factor, is known to be involved
via the increased expression of glycolytic enzymes.
HIF-1
activates the gene expression of glycolytic enzymes,
which are activated as compensation for mitochondrial
oxidation of fatty acids in the failing heart. Miyauchi
and colleagues identified by sequence analysis of
the 5’-flanking promoter region of ET-1 an HIF-1
recognition site in this region. Therefore, they hypothesized
that HIF-1
is involved in the increase in the myocardial expression
of the ET-1 gene in heart failure.
Work by Miyauchi and colleagues showed that HIF-1
transcriptionally activates ET-1 gene expression by
direct interaction with the predicted DNA binding
site in the 5’- promoter region of the ET-1 gene
in rat cardiomyocytes. HIF-1
mRNA and ET-1 mRNA in the failing heart increased
during the aggravation of heart failure in rats with
myocardial infarction and hamsters with cardiomyopathy.
In rat cultured myocytes treated with a mitochondrial
inhibitor, HIF-1
mRNA and ET-1 mRNA was markedly increased with activated
glycolysis. Furthermore, antisense oligonucleotide
for HIF-1
greatly inhibited this expression of ET-1 in cardiomyocytes.
Miyauchi concluded that ET-1 is an important aggravating
substance in heart failure. Various factors are involved
in the increase in ET-1 gene expression in the failing
heart. Their recent data has revealed a novel molecular
mechanisms for the upregulation of cardiac ET-1 in
heart failure in which impairment of cardiac energy
metabolism is involved in an increase in ET-1 expression
through HIF-1,
suggesting that induction of HIF-1
to stimulate glycolysis as an adaptation against impaired
energy metabolism alternatively causes an elevation
of cardiac ET-1 expression as a maladaptation, which
leads to aggravation of heart failure.
|
PAGE
TOP
|
Natriuretic
Peptides in Heart Failure |
|
The role of the natriuretic peptide system in the
basal condition was evaluated in the double knockout
mice model of GC-A, a receptor for natriuretic peptides,
and type I angiotensin II (Ang II) type receptor genes
by Saito and colleagues at the University of Kyoto.
Data from GC-A knockout mice clearly indicate that
the ANP-GC-A system functionally antagonized humoral
factors or pathways that lead to hypertension, left
ventricular hypertrophy (LVH) and cardiac fibrosis.
The actions of ANP and BNP are opposite those of
Ang II at every site of action. Thus to elucidate
the pathways for the functional antagonism of these
peptides, they generated a ANP-GC-A knockout mice.
Systolic blood pressure (SBP) was significantly higher
in the GC-A knockout mice than in the wild mice, consistent
with previous reports, while the SBP was significantly
lower in the AT1a knockout mice than in the wild type.
The SBP was significantly lower than that in the GC-A
knockout mice but significantly higher than that in
the AT1a knockout mice. The degree of reduction in
SBP between the wild type and the AT1a knockout mice
was similar to the degree of reduction from the GC-A
knockout to the double knockout mice—indicating
that the genetic blockade of the AT1a receptor similarly
decreases the SBP in the wild and GC-A knockout mice.
Thus, it is unlikely that Ang II contributed to blood
pressure elevation in the GC-A knockout mice. Their
work also showed that the genetic blockade of AT1a
was associated with a greater reduction in the ratio
of LV weight to body weight in the double knockout
mice, compared to the GC-A knockout and the AT1a knockout
mice models. Also they showed that the GC-A knockout
mice had more LV interstitial fibrosis than in the
wild type or the AT1a knockout mice. Less LV fibrosis
was observed in the double knockout mice than in the
GC-A knockout mice.
TGF-beta, collagen I and collagen III mRNA expression,
genes known to be responsible for fibrosis, was completely
blocked in the double knockout mice compared to the
AT1a knockout mice. The percent inhibition of SBP
was similar in the AT1a and double knockout mice.
The percent inhibition of LVW/BW and fibrosis was
significantly greater in the double knockout mice
than in the AT1a knockout mice. These results indicate
that the endogenous ANP-GC-A system plays an important
role in the functional inhibition of endogenous Ang-II-mediated
hypertrophy and fibrosis. However, the ANP-GC-A system
functionally blocks some pressor pathways other than
Ang II in the basal condition.
New roles for natriuretic peptides in the setting
of acute myocardial infarction using GC-A knockout
mice were also investigated by this group. Plasma
BNP is markedly elevated in patients who undergo PCI
after AMI. To determine the role of BNP or the ANP
system in the acute phase of MI, they developed a
mouse model of ischemia reperfusion using the GC-A
knockout mice. The ratio of the area of the infarct
area to the area at risk was about 60% in the wild
type and about 45% in the GC-A knockout mice. The
infarct size was significantly decreased by about
25% in the GC-A knockout mice at 2 days after reperfusion.
PMN infiltration in the GC-A knockout mice was much
smaller than in the wild type mice at 6 hours and
2 days after reperfusion. Increasing evidence shows
that the PMN infiltration into the infarct region
follows three steps, rolling migration, firm attachment,
and transmigration. The upregulation of P-selectin
in the endothelial cells play an important role in
binding to the PMN.
To understand the mechanism of increased infiltration
of PMN in the infarct region of the GC-A knockout
mice, they examined the endothelial expression of
P-selectin after ischemia reperfusion. P-selectin
was abundantly observed in the vessels in the perinecrotic
region of the viable myocardium near the infarction.
In contrast, few P-selectin cells are seen in the
GC-A knockout mice.
Activation of NF-kB, transcriptional factor that
upregulates the selectin gene in the endothelial cells,
is much weaker in the GC-A knockout mice than in the
wild type mice. P-selectin expression was increasingly
upregulated in human endothelial cells by the addition
of hydrogen peroxide, hydrogen peroxide plus ANP,
and even more with hydrogen peroxide plus ANP plus
an antagonist for GC-A.
|
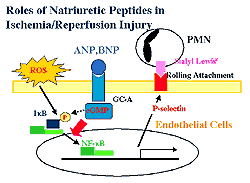 |
| Figure
1. The proposed mechanism of natriuretic peptide
system in ischemia perfusion. |
| Click
to enlarge |
|
Based on the data, these investigators proposed a
new role for GC-A in ischemia reperfusion. Radical
oxygen species generated by ischemia reperfusion phosphorylate
IkB, which leads the activation of NF-kB. Activated
NF-kB leads to the transcription of P-selectin and
then to PMN infiltration. GC-A or cyclic GMP potentially
regulates the ROS-induced NF-kB activation. These
findings provide a new insight into the role of the
natriuretic peptide system in ischemia reperfusion
(Figure 1).
|
PAGE
TOP
|
Cardiac
Ryanodine Receptor Alteration |
|
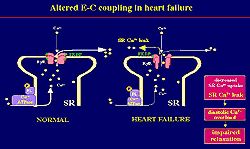
A decrease either in the activity or the protein
expression of the sarcoplasmic reticulum (SR) Ca2+-ATPase
is thought to be a major determinant in the pathogenesis
of cardiac dysfunction in heart failure.
A novel mechanism of cardiac dysfunction elucidated
by assessing the functional interaction of the FK
506-binding protein (FKBP12.6) with the cardiac ryanodine
receptor (RyR) in a canine model of pacing-induced
heart failure has been reported by Yano and colleagues
at Yamaguchi University and Tohoku University Graduate
School of Medicine. They showed that in heart failure
the stoichiometry of FK binding protein (FKBP) per
RyR was decreased. This partial loss of RyR-bound
FKBP12.6 seems to induce an instability in the properties
of the channel through a protein conformational change.
This leads to a prominent Ca2+ leak, which can cause
Ca2+ overload and hence diastolic and systolic dysfunction.
It has been recently demonstrated that FKBP are tightly
coupled with the RyR with a stoichiometry of 1 RyR
to 4 FKBP. Although FKBP appears to have a channel
stabilizing effect in skeletal muscle, controversy
remains regarding the role of cardiac FKBP on the
function of RyR. Thus, they assessed the role of the
interaction between FKBP and RyR in the pathogenesis
of heart failure.
Heart failure was created by chronic rapid right
ventricular pacing at a rate of 250 bpm for 3 weeks
in the canine model. After hemodynamic assessment,
the LV was isolated and the SR vesicles were purified.
The calcium leak from its initiation to its saturation
was measured in the presence of various concentrations
of FK506. It is known that FK506 specifically binds
to the FKBP and then dissociates FKBP from the RyR.
The hemodynamics showed that the LV, diastolic and
end systolic diameters and the LV end diastolic pressure
were increased in the heart failure model. There was
a decrease in fractional shortening. The +dP/dt pressure
of the LV pressure was decreased.
In normal SR, FK506 caused a dose-dependent calcium
leak that was similar to the significant conformational
change in RyR. In contrast, in the failing SR, a prominent
calcium leak was observed even in the absence of FK506,
and FK506 produced little or no further increase in
calcium leak and only a slight conformation change
in RyR.
Calcium release assay using stopped-flow apparatus
was performed. In normal SR vesicles, the addition
of FK506 decreased the rate of calcium release. However,
in heart failure, FK506 did not decrease the rate
of calcium release, which was already decreased compared
to in heart failure. These data indicate that some
of the FKBP dissociated by FK506 is already partially
lost in heart failure.
|
|
|
A polylysine-induced increase in ryanodine receptor
binding means deactivation of the ryanodine receptor.
In normal conditions, FK506 decreased the polarizing
induced enhancement of the ryanodine binding in a
dose-dependent fashion. However, in heart failure,
FK506 had no significant effect on polylysine-induced
increase in ryanodine receptor binding. In heart failure,
the Bmax of the FK506 was markedly decreased. However,
the Kd was unchanged. The stoichiometry of FKBP per
RyR was 1:3.6 in the normal heart, compared to 1:1.6
in heart failure. The protein expression of FKBP12.6
was significantly decreased in heart failure compared
to normal SR vesicles (p<0.05; Figure 2).
Yano concluded that in tachycardia-induced heart
failure, the rate of calcium release through RyR was
decreased in association with an abnormal calcium
leak through the RyR. This abnormality within this
protein is presumably caused by a partial loss of
RyR-bound FKBP12.6 and the resultant conformation
change in RyR. This abnormal channel gating in RyR
might possibly cause calcium overload and consequent
diastolic and systolic dysfunction.
|
PAGE
TOP
|
HB-EGF and
Metalloproteinase Inhibitors |
|
Heparin binding epidermal growth factor (HB-EGF)
was purified from macrophage-like cells U937 by Takashima
and colleagues at Osaka University. HB-EGF is a mitogen
for fibroblasts and vascular smooth muscle cells (VSMC)
but not for endothelial cells. HB-EGF is involved
in the pathogenesis of atherosclerosis. Neonatal and
adult cardiac muscle cells respond to both neurohumoral
and mechanical growth stimuli with a marked increase
in HB-EGF mRNA. The expression and protein synthesis
of HB-EGF was enhanced in the LV of spontaneous hypertensive
rats.
They investigated whether HB-EGF shed by metalloproteinases
plays an important role in the development of cardiac
hypertrophy. They hypothesized: That HB-EGF is involved
in cardiac hypertrophy. HB-EGF plays an important
role in the cardiac hypertrophic signaling by catecholamine,
angiotensin II and endothelin-1. The cellular mechanisms
by which these factors are linked with HB-EGF are
attributable to the shedding of HB-EGF via metalloproteinases.
Therefore, a metalloproteinase inhibitor attenuates
cardiac hypertrophy in vivo.
HB-EGF can phosphorylate the EGF receptor, and induce
ERK activation and cardiac hypertrophy, as shown by
this group. Further, the G-protein coupled receptor
(GPCR) agonists such as phenylephrine, angiotensin
II, and endothelin-1 can stimulate HB-EGF release,
and the EGF receptor phosphorylation can occur extracellularly,
induce ERK activation and cardiac hypertrophy.
The metalloproteinase inhibitor KB-R7785 can block
the GPCR agonist-induced hypertrophic stimulation.
They also showed that KB-R7785 can block EGFR phosphorylation
in rat neonatal cardiomyocytes. In a diabetic mouse
model of cardiac hypertrophy, KB-R7785 inhibited the
shedding of HB-EGF. In another experiment, they showed
that KB-R7785 attenuated cardiac hypertrophy induced
by aortic binding. LV thickness was also attenuated
by KB-R7785. The metalloprotienase inhibitor attenuates
cardiac hypertrophy in either diabetic or pressure
overloaded mice.
Takashima concluded that HB-EGF plays an important
role in the development of cardiac hypertrophy by
the shedding of HB-EGF via metalloproteinase. The
present results hint that the inhibition of the novel
pathway of metalloproteinase-HB-EGF signaling in cardiac
hypertrophy may be therapeutic in cardiac hypertrophy.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2001
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|