 |
|
|
|
| Cardiovascular Endocrinology and Metabolism: Progress and Promise |
|
Hanna B. Rubins
Minneapolis VA Medical Center,
Minneapolis, MN
Hiroaki Shimokawa
Kyushu University Graduate
School of Medical Sciences, Fukuoka, Japan
Seinosuke Kawashima
Kobe University Graduate
School of Medicine, Kobe, Japan
Koji Maemura
Graduate School of Medicine,
University of Tokyo, Tokyo, Japan |
|
|
|
|
|
|
|
|
Glycemic status, plasma insulin and cardiovascular disease |
|
A 2- to 6-fold increased risk of coronary heart disease
(CHD) is associated with the presence of three or
more of the risk factors high triglycerides, low HDL
cholesterol, abdominal obesity, hypertension and high
fasting glucose, which also identify the metabolic
syndrome. Insulin resistance, a key future of the
syndrome and related to obesity, particularly abdominal
obesity, can lead to glucose intolerance and diabetes.
The insulin resistant state can lead to hypertension,
through the increased sympathetic activation and the
increased re-absorption of sodium and water at the
renal tubular level that promotes endothelial dysfunction,
and to increased release of free fatty acids from
fat cells with reductions in plasma HDL and increases
in plasma triglycerides and small dense LDL.
The World Heath Organization defines overweight as
a body mass index (BMI) greater than 25, obesity greater
than 30, and morbid obesity greater than 40. In the
US, 50% of women and 60% of women were obese in year
2000. About 20% of both sexes are obese. Since 1991,
overweight has increased by 25% and obesity by 61%
in the US. In Japan, in the early 1990s, 20% of people
were overweight and 2-3% were obese. Diabetes prevalence
in the US increased from 4.9% in 1990 to 7.3% in 2000.
Undiagnosed diabetes increases the prevalence to 10%.
Diabetes prevalence is similar in Japan.
|
 |
| Figure1 |
 |
| Figure2 |
|
Fibrates exert a number of beneficial effects on
lipid metabolism, hemostasis and atherogenesis, and
reduce clinical events in people with atherogenic
dyslipidemia and the metabolic syndrome, as shown
by clinical trials. Many of the effects of fibrates
are mediated by their activity as a PPAR (peroximase
proliferatated activated receptor), whose characteristics
are outlined in Figure 1. Some effects of PPAR alpha
activation on atherosclerosis, an area of active research,
are shown in Figure 2. Activation of PPAR alpha induces
lipoprotein lipase (LPL) expression leading to increased
lipolysis of triglyceride-rich lipoproteins and a
decrease in triglycerides and increase in HDL. APO
C III expression is inhibited, leading to enhanced
LPL-mediated lipolysis, as well as APO I and APO II
expression, causing an increase in plasma HDL.
The fibrate gemfibrozil was associated with a 6%
increase in HDL, a 31% decrease in triglycerides and
no change in LDL cholesterol, compared to placebo,
in the VA HDL multicenter trial, stated Rubins of
the Minneapolis VA Medical Center. A 22% relative
risk reduction (RRR) for CHD death and nonfatal myocardial
infarction (MI) was observed (p<0.006). The trial
randomized 2,531 men less than 74 years of age with
CHD and LDL less than 140 and HDL less than 40 to
gemfibrozil or placebo and followed them 5 to 7 years.
A 28% RRR with gemfibrozil was seen in the nearly
one-half of patients with metabolic syndrome (p=0.001).
A more limited drug efficacy was seen in the patients
without the metabolic syndrome. A 37% RRR was seen
with gemfibrozil in patients with metabolic syndrome
and no diabetes. A 35% RRR with gemfibrozil was seen
in non-diabetic hyperinsulinemic patients (p=0.036).
Interestingly, this is the first report of a therapeutic
intervention that prevented clinical cardiovascular
events in non-diabetics with hyperinsulinemia.
|
|
Glitazones, insulin-sensitizing agents, improve insulin
stimulated glucose disposal, may promote insulin inhibition
of hepatic glucose production, and have effects on
lipid metabolism, hemostasis and atherogenesis. There
is no clinical trial evidence that these PPAR gamma
activators reduce cardiovascular events in the setting
of insulin resistance of metabolic syndrome. Favorable
effects of glitazones include inhibition of smooth
muscle cell migration and proliferation, inhibition
of secretion of inflammatory cytokines by activated
monocytes, inhibition of endothelial expression of
adhesion molecules, promotion of cholesterol efflux
from foam cells and apoptosis of activated macrophages.
Potential unfavorable effects are increased in macrophage
uptake of oxidized LDL and apoptosis of vascular endothelial
cells. Trial data is needed to determine whether the
favorable or unfavorable effects are dominant and
the efficacy of these agents on reducing cardiovascular
events.
|
PAGE
TOP
|
Identification of Endothelium-Derived Hyperpolarizing Factor |
|
Recent work by Shimokawa and colleagues has identified
that endothelium-derived hydrogen peroxide is an endothelium-derived
hyperpolarizing factor (EDHF). This identification
is thought to contribute to the evolving concept of
endothelium-derived relaxing factors (EDRF), because,
in addition to nitric oxide, another reactive oxygen
species served to maintain vascular function and homeostasis
in vitro and in vivo.
Endothelial cells play a key role in vascular homeostasis
by synthesizing and releasing vasodilating factors,
including prostacyclin, nitric oxide (NO) and EDHF.
Other important vasodilating factors include adrenomedullin
and C-type natriuretic peptide (CNP). The relative
contribution of prostacyclin, NO and EDHF to endothelium-dependent
relaxation (EDR) is markedly varied, depending on
the size of the blood vessels, and can be evaluated
experimentally by the inhibitory effect of indomethacin,
L-NNA and KCl. This group showed
that the contribution of EDHF increased as the vessel
size decreased. In contrast, NO seemed to play a major
role in large arteries. The same effects were shown
in isolated human mesenteric arteries, where EDHF
was important in causing bradykinin-induced EDR in
microvessels. That NO plays a major role in relatively
large arteries, whereas EDHF plays a major role in
small resistance arteries is thought to be true in
various species and vascular beds.
NO and EDHF synergistically
produced EDR in a relatively redundant manner in normal
blood vessels in a series of experimental and clinical
studies by these investigators. With increasing endothelial
injury caused by underlying risk factors, EDR was
progressively reduced. Both NO-mediated and EDHF-mediated
components were decreased with increased endothelial
injury, and were restored with improvement in the
endothelial injury. The calcium/calmodulin pathway
is required for the synthesis of both NO and EDHF.
Based on these lines of evidence, they hypothesized
that EDHF might be a non-NO factor derived from endothelial
NO synthase, which they investigated using eNOS-knockout
mice. In wild-type mice, EDR to acetylcholine (ACh)
was largely mediated by EDHF with some contribution
from NO. In contrast, in eNOS knockout mice, the EDR
to ACh was significantly reduced, but with some compensatory
augmentation by prostaglandin. Importantly, in the
knockout mice, the EDHF-mediated EDR was significantly
reduced. Endothelium dependent hyperpolarization in
response to ACh was significantly reduced in eNOS-knockout
mice compared to wild type mice. Vascular smooth muscle
relaxation responses to levkromaklium, a direct opener
of K channels, were normal in eNOS-knockout mice,
and were comparable in both strains. The response
to nitroprusside, a NO donor, was rather enhanced
in eNOS-knockout mice. These results appear to confirm
their hypothesis.
|
|
The non-NO factor derived from eNOS was then considered.
They examined the inhibitory effect of catalase, which
specifically converts hydrogen peroxide (H2O2)
into water and oxygen, to test their hypothesis that
superoxide is dismutated by SOD into H2O2,
which then may play a role as an endogenous EDHF.
These experiments demonstrated that 1) catalase markedly
inhibited EDHF-mediated responses, 2) exogenous H2O2
elicited similar relaxations and hyperpolarizations,
3) endothelial production of H2O2
was noted in wild-type mice but not in eNOS-knockout
mice in which EDHF-mediated responses were markedly
reduced. Other work has confirmed that H2O2
is an endogenous EDHF in porcine coronary microvessels
and in human mesenteric arteries, based on the finding
that catalase markedly inhibited the bradykinin-induced
EDHF-mediated relaxation and hyperpolarization. Also
confirmed was that exogenous H2O2
mimicked those EDHF-type responses. The results
of this work are summarized in Figure 3. The importance
of gap junction in the mechanism of EDHF-mediated
relaxation and hyperpolarizations has not been confirmed.
|
|
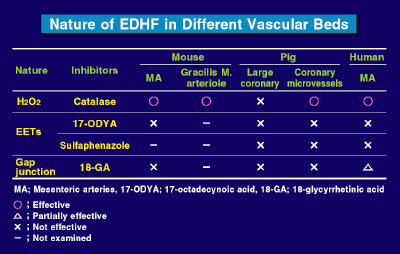 |
| Figure
3. Summary of EDHF effects in vascular beds studied
by Shimokawa and colleagues |
|
| Current work is focused on efforts to quantitatively
measure the production of H2O2 using
electron spin resonance (ESR) method. In the presence
of indomethacin and L-NNA, bradykinin
caused endothelium-dependent EDHF-mediated relaxations,
which were markedly augmented by Tiron. Catalase blocked
this augmentation, indicating that H2O2
mediated the effect of Tiron. In their experimental
series, the presence of normally functioning SOD is
essential. To examine the potential role of SOD as an
EDHF synthase, a series of experiments were performed.
In wild type mice, EDR to ACh was largely mediated by
EDHF with some contribution by NO, but in Cu/ZnSOD-knockout
mice, NO-mediated relaxation was surprisingly upregulated.
Importantly, EDHF-mediated components were markedly
reduced, indicating the important role of Cu/ZnSOD as
an endogenous EDHF synthase. In a working heart model,
coronary flow increased in both types of mice with bradykinin
stimulation. Catalase significantly inhibited this increase
in wild type mice, but not in the knockout mice, again
indicating that H2O2 is substantially
involved in the bradykinin-induced increase in coronary
flow.
In other work, coronary microvessels dilated in a
stepwise manner as perfusion pressure was decreased
from 70 mm Hg to 30 mmHg, representing autoregulatory
coronary vasodilation. L-NNA significantly
inhibited this autoregulatory relaxation, which was
interestingly further inhibited by catalase. This
indicates that both NO and H2O2 are
involved in coronary autoregulatory vasodilation in
vivo.
|
PAGE
TOP
|
The eNOS/NO system and maintenance of vascular structure |
|
Endothelial nitric oxide synthase (eNOS) produces
nitric oxide when L-arginine or its cofactor BH4 is
sufficient for the catalytic activity of eNOS. This
eNOS-derived NO acts to protect or maintain vascular
structural integrity. However, vascular tissue levels
of BH4 are relatively low under pathological conditions,
and the mechanisms for this are unclear. Activation
of eNOS leads to an "guncoupling of eNOS".
The uncoupled eNOS generates superoxide, which impairs
vascular function and promotes atherosclerosis. Hence,
eNOS has two different effects on vascular structural
integrity. In some cases these findings may suggest
caution about the recent therapeutic strategy of increasing
eNOS expression to treat cardiovascular disease. Seinosuke
Kawashima, M.D., of Kobe University, reviewed the
research conducted by he and his colleagues that support
this working hypothesis.
The role of the
eNOS/NO system in vivo using transgenic mice overexpressing
eNOS mainly at the level of the endothelium (eNOS-Tg)
was studied. In the eNOS-Tg mice, NO production and
cGMP concentration were increased.
The effects of eNOS-derived NO inhibit vascular
remodeling. In one model, intimal and medial thickening
occurred and lumen diameter decreased when the common
carotid artery was ligated just below the bifurcation
in wild type mice. In contrast, in the eNOS-Tg, intimal
and medial thickening was markedly attenuated. Vascular
remodeling was increased in both the wild type and
transgenic mice with L-NAME. Medial thickening of
small pulmonary arterioles produced by three weeks
of 10% hypoxia seen in wild type mice was attenuated
in eNOS-Tg mice. The ratio of muscular to non-muscularized
small pulmonary arterioles was increased with chronic
hypoxia in the wild type mice, but was attenuated
in the eNOS-Tg mice.
Overexpression of eNOS protects muscle tissue against
ischemia and reperfusion. This positive effect in
small vessels is at least in part mediated by the
maintenance of vascular permeability by eNOS-derived
NO. Superoxide production, detected by dehydroethydium
staining, was increased in response to 60 minutes
of femoral artery occlusion in the wild type animals,
but attenuated in the eNOS-Tg. Ischemia and reperfusion
increased vascular permeability in wild type mice
but was attenuated in ENOS-Tg mice, and tissue damage
was attenuated in ENOS-Tg mice.
eNOS-derived NO
inhibits inflammation, as shown by an experiment in
which marked lung inflammation in response to an injection
of LPS was seen in wild type mice but was significantly
less in eNOS-Tg mice. Ischemia-induced angiogenesis
is promoted by eNOS-derived NO. At 2 weeks, more collateral
vessels were seen in ischemic tissue of eNOS-Tg mice
compared to wild type mice in response to ligation
of the proximal portion of the femoral artery and
distal portion of the saphenous artery in the hindlimb.
In contrast to these positive effects, Xi and
colleagues showed that eNOS produces superoxide in
the absence of L-arginine or the cofactor BH4. BH4
improves depressed endothelium-dependent vasorelaxation
(EDR) in diabetes, hypertension, hyperlipidemia and
atherosclerosis in animal models and humans. BH4 improved
depressed EDR in isolated human coronary arteries
from patients with severe atherosclerosis. Superoxide
production by the endothelium of the aorta is increased
in apoE knockout mice, as shown by Harrison and colleagues.
Sepiapterin, a precursor of BH4, abates this superoxide
production.
A deficiency of eNOS promotes atherogenesis,
as shown by work in eNOS-knockout mice. In apoE knockout
mice crossed with eNOS-Tg mice and fed a high-cholesterol
Western diet, atherosclerotic lesion formation was
augmented. The marked increases in plasma lipid levels
were not modified by eNOS overexpression, while blood
pressure was lower in study mice, which may be involved
with the reduced lesion formation.
Further work confirmed the endothelium to be
the main production source for superoxide in the mice
aorta, as a result of eNOS overexpression. In aortas
from apoE knockout mice overexpressing eNOS, BH4 levels
were decreased and BH2, its oxidized form, increased
compared to control apoE knockout mice. In the presence
of hyperlipidemia, it is likely that BH4 levels are
insufficient for the overexpressed eNOS to produce
NO. Chronic BH4 treatment increased the BH4 content
and decreased the BH2 content in the aorta. Further,
superoxide production was decreased in the aorta of
apoE knockout mice overexpressing eNOS. In contrast,
NO production from aortas was increased with BH4 treatment.
The superoxide generated by eNOS in the presence of
reduced BH4 availability may promote atherosclerosis
lesion formation.
|
PAGE
TOP
|
Circadian Gene Expression in Vascular Endothelial Cells |
|
Work by Maemura and colleagues as the University
of Tokyo suggests that circadian output genes are
in part regulated by the central biological clock
with humoral factors or autonomous nervous system.
But, these genes may be directly regulated, at least
in part, by peripheral clocks in the cardiovascular
(CV) system, for example in vascular endothelial cells.
Prevention and treatment of CV diseases may benefit
from further understanding of this mechanism.
The biologic clock comprises transcriptional-translational
feedback loops. The CLOCK-BMAL1 heterodimer binds
to the E-box site in the promoter region of Per and
Cry genes and transactivates the Per and Cry genes.
The Per and Cry genes accumulate in the cytoplasm,
and translocate into the nucleus and turn off the
transcription of Per and Cry genes by CLOCK-BMAL1
heterodimers. This negative feedback loop occurs over
24 hours. CLOCK, BMAL1 and Per belong to the basic
helix-loop-helix (bHLH)/PAS domain transcription factor
family, whose features are shown in Figure 4.
|
 |
| Figure
4. Features of the basic helix-loop-helix (bHLH)/PAS
domain transcription factor family. |
|
|
CLIF, cycle like factor, was identified by this group
in their work to identify transcription factors important
for vascular endothelial function, using EPAS1 as
bait when screening the HUVEC cDNA library using the
yeast two-hybrid system. CLIF is a novel bHLH/PAS
protein that shares a high homology with Drosophila
CYCLE and its mammalian homologue BMAL1. Another research
group cloned the same gene and named it BMAL2. Maemura
and colleagues showed that CLIF interacts with EPAS1
and CLOCK, similar to BMAL1, and that a heterodimer
of EPAS1 and CLIF are important for hypoxic responses.
Work by this group shows that CLIF and CLOCK plays
a role in the biological clock. CLIF is expressed
in endothelial cells and neurons in the brain, including
the suprachiasmatic nucleus (SCN), the center of the
circadian clock, where it functions as a component.
It has been demonstrated that clock-related genes
are expressed in the SCN and in peripheral organs,
suggesting the existence of peripheral oscillators
in each organ. Unknown humoral factors synchronize
the peripheral and central clocks. Direct regulation
of physiologically important circadian output genes
may be a function of the peripheral clocks.
A peripheral clock in the CV system was demonstrated
by this group in mice on a 12-hour light and dark
cycle, in which BMAL1, Per2 and Cry1 exhibited circadian
variation in the heart, aorta and kidneys. They hypothesized
that the peripheral oscillator may exist in vascular
endothelial cells, since certain endothelial functions
in regulating vascular tone and fibrinolytic activity
show circadian variation. Northern blot analysis clearly
demonstrated the circadian oscillation of clock-related
gene expression in endothelial cells.
Then they hypothesized that CLOCK and CLIF may regulate
the circadian variation of PAI-1 gene expression in
endothelial cells. In mouse heart and kidney, circadian
variation of PAI-1 mRNA levels with peak evening expression
was demonstrated. This pattern is the opposite that
seen in humans, perhaps because rodents are nocturnal
and humans diurnal. Adenovirus-mediated gene transfer
showed that CLOCK overexpression results in a dose-dependent
increase in PAI-1 mRNA levels relative to the control
cells infected with a GFP-expressing adenovirus. A
further increase in PAI-1 mRNA levels occurred with
co-infection of an adenovirus expressing CLIF with
CLOCK. A 5-fold increase in PAI-1 promoter activity
occurred with cotransfection of CLOCK and CLIF using
human PAI-1 promoter/luciferase reporter plasmids.
|
|
Deletion analysis localized the responsive element
between -800 and -549. The two E-box sites, the consensus
binding sites of CLOCK and BMAL1, were shown to be
important for the CLOCK:CLIF transactivation of the
PAI-1 promoter, since mutation in either box decreased
transactivation and mutation in both abolished it.
The CLOCK:CLIF heterodimer was shown to transactivate
the PAI-1 promoter by directly binding to the E-boxes.
Per2 and Cry1, the negative components of the biological
clock, were shown to suppress the CLOCK:CLIF-mediated
transcription of PAI-1, resulting in the circadian
oscillation of PAI-1 gene expression.
Their results suggest the presence of a peripheral
oscillator in the vascular endothelial cells, comprised
of CLOCK, CLIF, Per and Cry, which may regulate the
circadian expression of PAI-1 genes as a circadian
output gene (Figure 5). In cultured primary rat cardiomyocytes,
the rhythmic expression of BMAL1, Per2 and Cry 1 was
seen. Figure 6 summarizes their research findings.
|
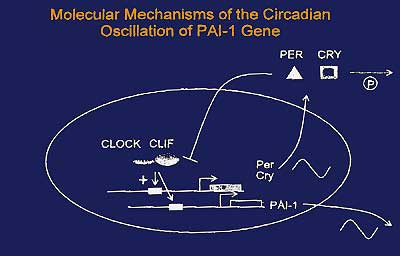 |
| Figure
5. Mechanisms by which a peripheral oscillator
in the vascular endothelial cells may regulate
the circadian expression of PAI-1 genes. |
 |
| Figure
6. Summary of research findings by Maemura and
colleagues |
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2002
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|