|
|
|
|
| New Alternative Therapies for Refractory Heart Failure |
|
Seiji Takashima
Osaka University School of
Medicine, Osaka, Japan
Hiroshi Okamoto
Hokkaido University Graduate
School of Medicine, Sapporo, Japan
Masafumi Yano
Yamaguchi University School
of Medicine, Ube, Japan
Shinji Tomita
National Cardiovascular Center
Research Institute, Suita, Japan |
|
|
|
|
|
|
|
|
Inhibition of cardiac hypertrophy by HB-EGF |
|
Biochemical studies performed by this research group
with heparin-binding EGF in refractory heart failure
were reviewed by Seiji Takashima, MD, of Osaka University
School of Medicine. G-protein coupled receptor (GPCR)
agonists induce cardiac hypertrophy and cause shedding
of HB-EGF via metalloproteinase activation, leading
to transactivation of the epidermal growth factor.
In mice with cardiac hypertrophy due to aortic banding
or GPCR agonists, KB-R7785 inhibited HB-EGF shedding
and attenuated cardiac hypertrophic changes. HB-EGF
and its signal transduction are specific important
molecules among the EGF family ligands for cardiac
hypertrophy.
|
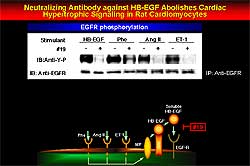 |
| Figure
1. HB-EGF shedding is inhibited and hypertrophic
changes attenuated by KB-R7785 in the rat model
of cardiac hypertrophy. Antibody 19 blocked any
effect by angiotensin II, endothelin-1 and phenylephrine,
showing this effect to be specific to HB-EGF. |
| Click
to enlarge |
|
Work in KKAY mice led to the hypothesis that KB-R7785,
a new inhibitor of metalloproteinase, attenuates cardiac
hypertrophy by preventing shedding of HB-EGF shedding.
Further work showed that KB-R7785 inhibited HB-EGF
shedding and attenuated hypertrophic changes in rats
with cardiac hypertrophy. Using the new antibody number
19, they showed that the effect is specific to HB-EGF,
since any effect by angiotensin II, endothelin-1 and
phenylephrine (PE) were blocked (Figure 1).
Cloning the shedding enzyme of HB-EGF showed that
it bound to KB-R7785 with high affinity. The activated
domain was used for hybrid screening with the heart
cDNA library, from which ADAM (a disintegrin and metalloproteinase)
12 was identified. A mutant strain of metalloproteinase
domain mutant 1 was prepared and compared to wild
type ADAM12. KB-R7785 inhibited shedding in MOCK and
the mutant strain, while increased shedding was seen
in the wild type. Then, transactivation was studied
in control, wild type and mutant rat cardiomyocytes.
Transactivation with PE was unchanged in the control
cardiomyocytes, but inhibition occurred in the dominant
negative mutant cells. Blockade did not occur with
point mutation. Then to determine the target of KB-R7785,
they showed that Biotinylated KB-R7785 bound to wild
type ADAM12 within the ADAM family, but did not bind
to ADAM12 mutation.
|
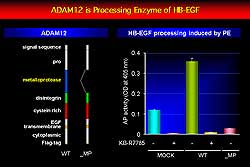 |
| Figure
2. ADAM 12 is the shedding enzyme for HB-EGF shedding
in cardiomyocytes. |
| Click
to enlarge |
|
Together, this work identified ADAM12 as the shedding
enzyme for HB-EGF shedding in cardiomyocytes (Figure
2), that ADAM12 binds to KBR-7785, and that KB-R7785
specifically inhibits HB-EGF shedding. In the aortic
banding mouse model, KB-R7785 is easy to administer
without any hemodynamic changes. KB-R7785 attenuated
cardiac hypertrophy caused by GPCR agonists after
7 days of PE or 14 days of angiotensin II.
HB-EGF binding seems to be involved with worsening
hypertrophy and HB-EGF signaling might be involved
with the alteration of cardiac function. Inhibiting
ADAM12 or HB-EGF, perhaps with angiotensin II or PE,
may be a beneficial therapeutic approach. However,
more than one pathway could be involved in the process
of developing heart failure, limiting the efficacy
of these approaches, and further study is needed to
assess these possibilities.
|
PAGE
TOP
|
Immunosuppressive Therapy in Refractory Heart Failure: Blockade of T Cell
Costimulatory Signals |
|
A novel view of inflammatory mechanisms potentially
involved in the development of heart failure holds
that various stimuli, including autoimmunity, infection
and mechanical overload and ischemia, induce production
of inflammatory cytokines, which negatively influence
contractility and contribute to the remodeling process
in the failing myocardium, resulting in heart failure.
Hiroshi Okamoto, MD, Hokkaido University Graduate
School of Medicine, Sapporo, Japan reviewed work by
his and other research groups of immunosuppressive
therapy.
Anti-TNF alpha therapy causes a significant dose-dependent
decrease in ejection fraction, improved left ventricular
end-diastolic and end-systolic volumes and mass, and
functional status. Immunomodulation in heart failure
remains controversial and large-scale trials are needed
to confirm results in smaller trials evaluating anti-cytokine,
immunoabsorption, immunosuppressive and intravenous
immunoglobulin therapies.
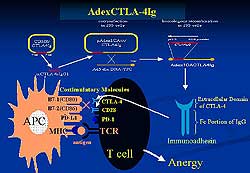
The blockade of costimulatory molecules as an important
strategy for treating autoimmune heart disease is
indicated by work Seiko and colleagues showing that
antigen-specific T-cells expressing cytotoxic lymphocytes
(CTL) and natural killer (NK) cells infiltrated the
heart concomitantly with B7-1, B7-2 and CD-40 in cardiac
myocytes in patients with dilated cardiomyopathy (DCM)
and myocarditis.
T-cells require two distinct signals for full activation.
The first is provided by the engagement of the T-cell
receptor (TCR) with MHC on antigen-presenting cells
(APCs) and the second signal by engagement of one
or more T-cell surface receptor with their ligands
on APCs. CD-28, cytotoxic lymphocyte antigen 4 (CDA4)
and B7 play major roles in T-cell activation. CTLA-4
effectively inhibits this ligation as immunoadhesion
and induces antigen-specific T-cell responses.
|
|
|
A recombinant adenovirus AdexCTLA-4IgG was constructed
by Okamoto and colleagues due to the limited half-life
of CTLA-4 and difficulty maintaining sufficient cell
concentration (Figure 3). Characteristics of AdexCTLA-4IgG
include a dose-dependent cell concentration maintained
at a sufficient dose 180 days after only one intravenous
(IV) administration. AdexCTLA-4IgG-mediated gene expression
occurs mainly in liver cells and maintains a sufficient
serum concentration in vivo.
|
|
Host immune responses have limited the use of recombinant
adenoviruses for gene therapy, in part due to cytotoxic
lymphocyte activation. In the liver, there is little
if any inflammatory response after AdexCTLA-4IgG administration.
Complete blockade of anti-viral antibody production
is achieved with AdexCTLA-4IgG administration. The
serum concentration of CTLA-4IgG was significantly
elevated after the second administration in mice treated
with AdexCTLA-4IgG. However, CTLA-4IgG is not detected
in mice pre-treated with AdexACE because of antibody
production. Thus, AdexCTLA-4IgG alone can completely
prevent anti-viral antibody production in vivo, leading
to a persistent and efficient serum concentration
of CTLA-4IgG.
The effects of AdexCTLA-4IgG before and after the
onset of experimental autoimmune myocarditis (EAM)
was studied by Okamoto and colleagues. Although EAM
is transferable by activated T-cells, the effect of
T-cell activation blockade on the prevention of EAM
was unknown. In the AdexLacZ-treated rats, 25% of
EAM rats died of severe myocarditis and heart failure,
while none died in the AdexCTLA-4IgG-treated groups.
Expression of the co-stimulatory molecules B7-1, B7-2,
CD40, CD28, and GAPDH was enhanced in the AdexLacZ
treated EAM rats. Enhanced expression of B7-1, B7-2
and CD40 was observed on cardiac myocytes on immunohistochemical
staining. Thus, myocytes as APCs may play a critical
role in the engagement with T-cells.
All rats treated with AdexLacZ showed discoloration
of the surface and cardiac enlargement and developed
typical autoimmune regions composed of inflammatory
cells. The AdexCTLA-4IgG-treated rats showed microscopic
abnormalities, with little infiltration of inflammatory
cells in the myocardium. On day 14, minimal myocarditis
was observed. The heart weight to body weight ratio
in the AdexCTLA-4IgG-treated rats was significantly
lower than in AdexLacZ-treated rats, and nearly equivalent
to that in normal rats. These findings indicate the
benefits of adenovirus-mediated co-stimulatory signal
blockade after the onset of myocarditis.
|
 |
| Figure
4. AdexCTLA-4IgG suppressed cytokine activation
and the onset and progression of experimental
autoimmune myocarditis in the rat model. |
| Click
to enlarge |
 |
| Figure
5. AdexCTLA-4IgG pre-treatment reduced the size
of the infarcted and affected area in a model
of ischemic/reperfusion injury after cardiac transplantation,
while pre-treatment with AdexLacZ was associated
with a novel 40% infarcted area and a novel 30%
affected area. |
| Click
to enlarge |
|
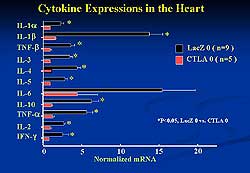
Cytokine expression in the EAM rats is shown in Figure
4. AdexCTLA-4IgG almost completely inhibited cytokine
expression, in contrast to AdexLacZ. The onset and
progression of EAM was prevented by AdexCTLA-4IgG
concomitantly with the suppression of cytokine activation.
Ischemic/reperfusion injury after cardiac transplantation
was evaluated using an in vivo system in which
AdexCTLA-4IgG or AdexLacZ was injected via a recipient
vein 5 days prior to transplantation. Four hours of
ischemia after pre-treatment with AdexLacZ resulted
in a novel 40% infarcted area and a novel 30% affected
area evaluated by extent of inflammatory cell infiltration.
Pre-treatment with AdexCTLA-4IgG reduced the size
of the infarcted and affected area, as shown in Figure
5.
In the absence of AdexCTLA-4IgG treatment, there
was severe inflammatory cell infiltration and slight
but significant myocyte apoptosis occurred. Pre-treatment
with AdexCTLA-4IgG suppressed inflammation and the
number of TUNEL-positive cells. The mechanism by which
AdexCTLA-4IgG protects against reperfusion injury
could involve inhibition of the immune response and
increased resistance of cardiomyocytes to apoptosis.
In humans unable to mount immune responses against
various bacteria and bio-insults after AdexCTLA-4IgG
administration, long-lasting CTLA-4IgG expression
may be problematic. A new adenovirus vector containing
the AdexCre system was constructed, which terminated
expression of AdexCTLA-4IgG. Hepatic CTLA-4IgG production
was stopped. This new vector enabled termination of
in vivo gene expression at the desired time.
FK506, CsA and anti-cytokines impaired T-cell receptor
signal transduction leading to pan-T-cell suppression,
including na_ve T-cells and antigen-specific T-cells.
In contrast, CTLA-4IgG specifically blocked the costimulatory
signaling between antigen-treated cells and T-cells
only when antigen is present. Moreover, AdexCTLA-4IgG
caused persistent and efficient expression in vivo
for a long period that was terminated at the desired
time, but it has not been clinically evaluated. Potential
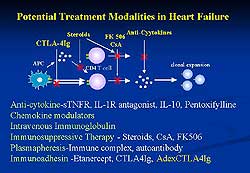
immunomodulator therapies including AdexCTLA-4IgG
are shown in Figure 6 and possible treatment modalities
of AdexCTLA-4IgG are summarized in Figure 7.
|
|
 |
| Figure
7 |
|
PAGE
TOP
|
Stabilization of calcium channel release |
|
Research by Yano and colleagues demonstrates that
excess beta-stimulation induces PKA-mediated hyperphosphorylation,
inducing the dissociation of FK-binding proteins (FKBP)
from the ryanodine receptor (RyR), conformational
change of RyR, and then Ca2+ leak. This
series of events within the RyR might cause Ca2+ overload,
and in turn cardiac dysfunction and heart failure.
Importantly, they demonstrated that FKBP-mediated
stabilization by JTV519 or a beta-blocker prevents
development of heart failure. Their results may represent
new strategies to prevent and treat heart failure.
Stabilizing the Ca2+ release channel of
the cardiac sarcoplasmic reticulum, often called the
ryanodine receptor, addresses a chief pathogenic mechanism
for various types of dysfunctions in heart failure.
FKBP are tightly coupled with the RyR and have a channel
stabilizing effect in skeletal and cardiac muscles.
A partial loss of RyR-bound FKBP12.6 causing a prominent
abnormal Ca2+ leak leading to conformational
changes was demonstrated by this group in a dog model
of pacing-induced heart failure. Presumably, this
abnormal Ca2+ leak causes Ca2+
overload, and hence results in diastolic and systolic
dysfunctions.
PKA-mediated hyper-phosphorylation of RyR causes
dissociation of FKBP12.6 from RyR, in turn causing
an increased sensitivity to Ca2+-induced
activation and defects in channel functions, as demonstrated
by Marx and colleagues. This suggests that failing
hearts lack normal FKBP12.6-mediated channel regulation.
Hence this group assessed FKBP12.6-mediated stabilization
of RyR as a novel therapeutic strategy for heart failure.
In an experiment model of heart failure produced
by 4 weeks of rapid RV pacing, FK506 binds to FKBP
and dissociates it from RyR, inducing a prominent
Ca2+ leak in normal SR. In failing SR,
spontaneous Ca2+ leak occurs. A new benzothiazepine
derivative, the cardioprotective agent JTV519, which
inhibits intracellular Ca2+ overload due
to ischemia-reperfusion, completely inhibited the
Ca2+ leak.
In normal SR, FK506 induced an increase in MCA (mechylcoumarin
acetate) fluorescence, which was inhibited by JTV519.
In failing SR, there was virtually no further increase
in MCA fluorescence. Interestingly, JTV519 decreased
the level of MCA fluorescence, suggesting that JTV519
restores the normal conformational state of RyR in
failing SR. The MCA fluorescent change was much faster
than the Ca2+ leak, indicating the MCA
fluorescent change precedes Ca2+ leak.
Chronic administration of JTV519 decreased end-diastolic
pressure (EDP), tended to increase +dP/dt of LVP,
shortened Tau, and strikingly reduced the LV chamber
size, compared to failing dogs with 4 weeks of right
ventricular pacing. Treatment with JTV519 reduced
left ventricular (LV) chamber size and attenuated
the development of LV remodeling.
The ability of beta-blockade to prevent heart failure
by restoring FKBP12.6-mediated stabilization of RyR
was then studied. Propranolol, 0.05mg/kg/day for chronic
administration, decreased heart rate, but had no effect
on LV pressure and contractility. This dose of propranolol
seemed to exert a negative chronotropic action, but
not a negative inotropic action. A higher dose caused
death due to heart failure in the canine model. Chronic
infusion of propranolol decreased LVEDP, shortened
Tau, reduced LV size and increased fractional shortening.
Under normal conditions, Ca2+ leak was
induced by the addition of FK506. In failing SR, spontaneous
Ca2+ leak was observed, and FK506 had no
further effect on the Ca2+ leak. In the
SR taken from propranolol-treated dogs, there was
virtually no spontaneous Ca2+ leak, and
FK506-induced Ca2+ leak appeared like in
normal SR. MCA fluorescence change was induced by
FK506 in normal SR, while no change was observed in
failing SR, as conformational change might have already
occurred in failing SR. In propranolol-treated SR,
FK506-induced MCA fluorescent change was partially
restored. Back phosphorylation was lower in failing
RyR than in normal RyR, indicating that RyR was hyperphosphorylated
in failing SR, whereas it was restored towards normal
in propranolol-treated SR. Ca2+ uptake
and expression of Ca2+ ATPase were decreased
similarly, regardless of the treatment with propranolol,
in both groups.
Marks and colleagues also demonstrated that PKA-mediated
hyperphosphorylation was restored by treatment with
the beta1-selective blocker, metoprolol, in the same
canine model. They further showed that the expression
of phospatase in RyR, that is PP2A and PP1, was decreased
in heart failure. This downregulation of PP1 and PP2A
may contribute to the hyperphosphorylation of RyR.
But, they were restored to normal in metoprolol-treated
heart. Consistent with the finding by Yano and colleagues,
the expression of FKBP12.6 was decreased in heart
failure and was reversed with metoprolol.
Several proposed mechanisms for the improved cardiac
function in chronic heart failure with beta blockers,
include inhibition of beta-adrenergic receptor kinase,
upregulation of SERCA and inhibition of metalloproteinase.
The inhibition of hyperphosphorylation of RyR and
the subsequent suppression of Ca2+ leak
might also lead to an improvement of cardiac function
through an inhibition of intracellular Ca2+
overload.
|
PAGE
TOP
|
Cell-based Therapy for End-Stage Heart Failure |
|
The concept of transplantation of exogenous cells
to compensate for myocyte loss as an alternative treatment
for end-stage heart failure has been explored since
1992. Allogenic, xenogenic and autologous cell sources
have been explored, with autologous sources showing
the most promise. Work by Tomita and colleagues sand
others in cell-based therapy was reviewed by Shinji
Tomita, National Cardiovascular Center Research Institute,
Suita, Japan.
In a landmark study by Soonpaa and colleagues, fetal
cardiomyocyte transplantation enabled synchronous
myocardial contraction due to formation of Gap junction.
Concerns about immunorejection of fetal cardiomyocytes
led to study of autologous cell transplantation. Skeletal
myoblast transplantation has been studied experimentally
and clinically, with Menasche and colleagues in France
showing their viability. However, since cell transplantation
is done as part of a revascularization procedure of
the coronary artery, determining which is more effective
is difficult. The detailed analysis of the ten completed
cases is awaited.
Bone marrow cells (BMC) for transplantation are strong
candidates in heart failure, particularly mesenchymal
stem cells. Myogenic cells were differentiated in
a study by Wakitani and colleagues. Beating cardiomyogenic
(CMG) cells were developed from BMC by Makino and
colleagues of Keio University in Tokyo, Japan. Tomita
and colleagues used BMC for autologous transplantation
and reported detectable Troponin I and improved heart
function. Bone marrow stroma cells were transplanted
into a porcine model by Tomita and colleagues after
which cardiomyocyte tissue was detectable. One month
after post-myocardial infarction and stroma cell transplantation,
perfusion and wall thickening were improved.
Human mesenchymal stem cell transplantation into
fetal sheep early in gestation resulted in site-specific
differentiation in work by Liechty and colleagues.
In terms of which part of the BMC are responsible
for this improved heart function, Orlic and colleagues
demonstrated that transplantation of Lin c-Kit+
cell after myocardial infarction resulted in good
differentiation and improved heart function.
|
 |
| Figure
8 |
 |
| Figure
9 |
|
Angiogenesis with endothelial progenitor cells (EPC)
and bone marrow mononuclear cell has been studied.
Clinical trials of BMC transplantation examining the
use of adult stem cells, bone marrow mononuclear cells,
EPC, mesenchymal stem cells and cardiomyocytes are
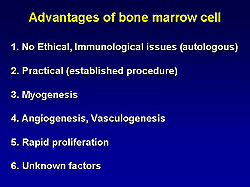
underway in Japan. Advantages of BMC are shown in
Figure 8.
An in vivo study using a co-culture system
in which cardiomyocytes were the host and green fluorescent
protein expressing mouse bone marrow cell (GFP-BMC)
the donor demonstrated synchronous contraction in
the cardiomyocytes after two days. Tomita and colleagues
also showed myosin heavy chain expression from day
1 and connexin43 and ANP from day 2 and cardiac-specific
Troponin I after day 4.
Clinically, cell-based therapy has been used in patients
waiting heart transplantation in Japan. Six patients
on a left ventricular assist device recovered and
were weaned form the device. This may change the environment
for heart transplantation. However, a number of issues
must be resolved for clinical application (Figure
9). Side effects such as arrhythmia must be addressed.
Clarification of the mechanism and the best type of
human cell is required. Improved evaluation methods
in humans are needed, new cell sources must be identified,
and facilities that comply with good manufacturing
principles are required.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2002
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|