|
|
|
|
| Cardiovascular Neurohumoral System:
Novel Aspect of Angiotensin and Aldosterone Receptor |
|
|
Angiotensin II as an Inflammatory Mediator
Akira Matsumori
Kyoto University Graduate School of Medicine, Kyoto, Japan
Role of Angiotensin II Type 1 Receptor in the Regulation of Angiogenesis
Toyoaki Murohara
Nagoya University Graduate School of Medicine, Nagoya, Japan
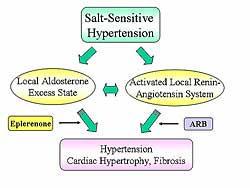
Local
Renin-Angiotensin System Induces Ventricular
Remodeling through Oxidant Stress-mediated Breakdown
of Extracellular Matrix in Rats with Salt-Sensitive
Hypertension
Yasuki Kihara
Kyoto University Graduate
School of Medicine, Kyoto, Japan
Effect
of Eplerenone, a Novel Selective Aldosterone
Receptor Antagonist in Salt-Sensitive Hypertension
Yoshiyu Takeda
Kanazawa University, Kanazawa,
Japan
Blockade
of Cardiac Aldosterone Production as a New Therapeutic
Strategy of Heart Failure
Michiro Yoshimura
Kumamoto University, Kumamoto,
Japan
|
|
|
|
|
Angiotensin
II as an Inflammatory Mediator
Akira Matsumori
Kyoto University Graduate School
of Medicine, Kyoto, Japan
|
|
Heart failure is an inflammatory disease and Angiotensin
II (Ang II) plays a role as an inflammatory mediator.
This is a new hypothesis by this group of investigators,
and the speaker presented data from their research
to support their theory.
Inflammation has been shown by recent studies to
be an important aspect of cardiovascular (CV) disease
and infection. A viral infection, hypertension, or
myocardial infarction (MI) induces an inflammatory
response in the heart and this inflammation comprises
cell infiltration, such as macrophages, mast cells,
and lymphocytes, which produce cytokines and may lead
to the development of heart failure.
Mast cells induce allergic inflammation and are
activated by IgE, thrombin, and neuropeptides such
as Substance P and complements. Activated mast cells
release granules that increase histamine, heparin,
cytokines, growth factors, metalloproteinases, and
proteases. These factors play an important role in
angiogenesis, inflammation, and fibrosis, which in
turn play a critical role in heart failure and cardiomyopathies.
|
|
Evidence from their research
Mast-cell deficient mice did not develop heart failure
in pressure overload-induced hypertrophy by aortic
banding, in experiments by this group. Chamber dilation
and decreased systolic function were seen in wild-type
(WT) mice, but not in mast-cell deficient mice (WW).
In the murine model of dilated cardiomyopathy (DCM)
induced by the encephalomyocarditis viral (EMCV) infection,
heart failure developed after 2 weeks of infection
and dilatation and hypertrophy developed 3 months
after infection. In this animal model, mast cell-mediators,
such as mast cell protease 4 (mMCP-4) and mMCP-5,
both chymases, and mMCP-7, a tryptase, are upregulated
in the heart failure stage with myocarditis. Membrane
type MMP-2 (MT-MMP-2), MMP-9, and Type I procollagen
are upregulated in the heart failure stage with myocarditis.
In further studies, they showed that survival was
better in the WW mast cell-deficient mice than in
the WT mice (about 85% vs about 35%), and there was
less cell infiltration (1.0 vs 0.1 histologic score,
respectively; p<0.01) and myocardial necrosis (0.7
vs 0.1 histologic score, respectively; p<0.01).
In other studies, Ang II was present in human mast
cells (HMC), which are already preformed in the cytoplasm
of mast cells. In Ang II HMC lines, mast cells from
the human lung and umbilical cord blood contain Ang
II. Stimulation of the mast cells by calcitonin, a
gene-related peptide (CGRP), caused Ang II release
into the supernatant in the cultured mast cells. CGRP
induced angiotensinogen expression in these mast cell
lines. The genes of angiotensinogen and renin were
found, but ACE was not found, in these cell lines.
Therefore, it is likely that in this mast cell line,
Ang II was formed by chymase.
Recent studies by other investigators have shown
that Ang II induces gene transcription through cell-type-dependent
effects on NF-kB, that NF-kB is activated through
AT1 and AT2 receptors in vascular smooth muscle cells,
and the transcription factor for NF-kB is necessary
for the upregulation of type 1 angiotensin II receptor
mRNA in rat cardiac fibroblasts treated with tumor
necrosis factor- and interleukin 1ß.
and interleukin 1ß.
|
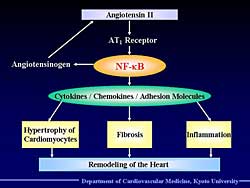 |
| Figure
1. The process of how angiotensin II leads to
remodeling. |
| Click
to enlarge |
|
Angiotensin II activates NF-kB through the AT1 receptor,
and this activation in hepatocytes probably induces
angiotensinogen, and this creates a positive feedback
circuit and can lead to remodeling of the heart (Figure
1).
As shown by this group, Ang II injection activates
NF-kB in WT mice, but does not activate the AT1 receptor
in knockout mice. Also, circulating Ang II is elevated
in the heart failure stage in the animal model of
heart failure due to myocarditis, and the presence
of circulating Ang II precedes heart failure. Ang
II and cytokines become present at about the same
time.
Improved survival in AT1-receptor deficient mice
infected with the EMCV myocarditis, compared to WT
mice (40% vs 20%, respectively; p<0.05) was found
in other experiments. Further, NF-kB activity was
increased in the WT mice but not in the AT-1 receptor
knockout mice. TNF-
and IL-1ß expression was enhanced in the WT
mice, but was lower in the AT-1 receptor knockout.
Also, iNOS expression was decreased in the knockout
mice. Candesartan inhibited expression of TNF-,
IL-1ß, and NF-kB.
Reports from other investigators showed that a mineralocorticoid
receptor activated NF-kB in double transgenic rats
for human renin and angiotensinogen genes. Also, that
aldosterone induces inflammation in the vasculature,
heart, and kidneys. These data indicate that the renin-angiotensin-aldosterone
system may play a role in inflammation in heart failure.
|
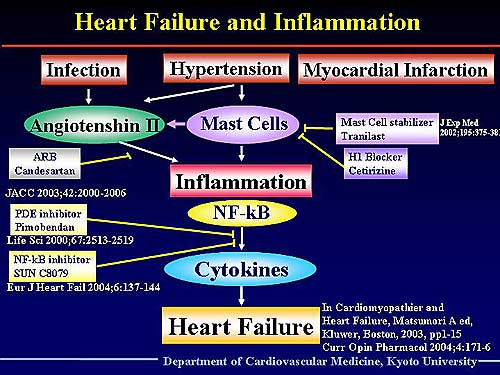 |
| Figure
2. Schematic of their proposed hypothesis of the
role of inflammation in the transition to heart
failure. |
| Click
to enlarge |
|
Thus, they hypothesize that in addition to the direct
myocytolysis and immunological damage to cytotoxic
lymphocytes and cytokines, the EMCV infection also
induces mast cell recruitment to the heart and might
induce mast cell degranulation, which induces mast
cell proteases and release of cytokines. This may
play an important role in myocardial damage, inflammation,
and remodeling.
Figure
2 illustrates their hypothesis. Infection, hypertension,
or MI induces inflammation in the heart. Cytokines
play a major role in the development of heart failure.
This group has shown that the PDE inhibitor pimobendan
and a new NF-kB inhibitor (SUNC8079) prevented the
development of heart failure by inhibiting the production
of cytokines. Ang II also induces inflammation and
the ARB candesartan prevented the development of inflammation
in heart failure. Mast cells play a very important
role in inducing Ang II and inflammation, and the
mast cell stabilizer tranilast and the histamine 1
blocker cetirizine prevents this inflammation and
heart failure. Anti-inflammatory therapy is a promising
future treatment of heart failure.
|
PAGE
TOP
|
Role
of Angiotensin II Type 1 Receptor in the Regulation
of Angiogenesis
Toyoaki Murohara
Nagoya University Graduate School
of Medicine, Nagoya, Japan
|
|
The regulation of angiogenesis through the Angiotensin
(Ang) II Type 1 receptor is a newly understood mechanism.
Work by Murohara and colleagues presented in this
lecture showed that the Ang II type 1 receptor pathway
functions as a promoter of the ischemia-induced neovascularization
in vivo by supporting inflammatory mononuclear cell
infiltration. Further, angiogenesis was mediated by
the Ang II type 1a receptor in both hindlimb ischemia
and tumor implantation models in mice.
The enhanced angiogenesis was likely mediated at least
in part by Ang II-induced inflammatory cell infiltration,
which can release a variety of angiogenic cytokines.
|
|
Study background
Previous research by other investigators showed
that an ACE inhibitor can protect against the risk
of cancer in patients with hypertension, and basic
research showed that the ACE inhibitor captopril inhibited
the basic VEGF-induced angiogenesis in a rat colony
micropocket assay, but other work showed an ACE inhibitor
increased angiogenesis in the hind leg ischemia model.
ACE inhibitors block the conversion of Ang I to
Ang II and inhibit the breakdown of bradykinin. The
increased bradykinin stimulates endothelial cells
to release nitric oxide and prostacyclin, molecules
known to modulate angiogenesis. Thus, ACE inhibitors
have a limited ability to regulate angiogenesis because
of the bradykinin-nitric oxide pathway.
Therefore, this group examined the role of the AT1
receptor, a major effective receptor for vascular
or cardiac tissue by Ang II in 1) ischemia-induced
angiogenesis (mouse hindlimb ischemia), and 2) tumor
angiogenesis (mouse tumor implantation). The Ang II
type 1a receptor (AT1a-/-) knockout mice model, with
reduced systemic blood pressure, was used.
|
|
Ischemia-induced angiogenesis
A similar marked reduction of blood perfusion in
the ischemic limb, after resection of the left femoral
artery and veins, was found in both the AT1a-/- knockout
mice (KO) model and wild type (WT) mice. However,
the recovery of blood flow in the ischemic hindlimb
was much greater in the AT1a-/- KO mice than in the
WT mice. The recovery of blood perfusion in the ischemic
limb was much greater in the WT mice than in the KO
mice. These data suggest that angiogenesis after hindlimb
ischemia might be reduced in the AT1a-/- KO mice.
Micovascular angiogenesis by capillary density and
arteriogenesis identified by angiography and functional
blood flow were all reduced in the KO mice compared
to WT mice. On day 14, the angiographic score was
reduced in the KO mice compared to the WT mice. In
isolated ischemic skeletal muscle immunostained with
anti-CD31 monoclonal antibody, at day 14 the capillary
density was decreased in the KO mice compared to the
WT mice (about 18/field vs 28/field, respectively).
Hydralazine, given to the WT mice to reduce blood
pressure to a level similar to the KO mice, had no
effect on angiogensis or blood flow recovery in WT
mice. However, after treatment with the AT receptor
blocker TCV-116, the ischemic/normal hindlimb blood
flow ratio in the WT mice was more similar to the
KO mice.
Ang II induces an inflammatory response, and plays
an important role in the infiltration of macrophages
or lymphocytes in the ischemic hindlimb. In the KO
mice, infiltrated T-lymphocytes or macrophages were
reduced. Interestingly, the infiltrated cells intensely
expressed VEGF, on double immunofluorescence staining.
Thus, VEGF released by the infiltrated inflammatory
cells might contribute to angiogenesis in ischemic
tissue. Therefore, the ischemia-induced angiogenesis
was markedly reduced in the KO mice.
The implantation of WT mast-derived mononuclear
cells directly into ischemic tissue of KO mice restored
the ischemic/normal laser Doppler blood flow ratio
after hindlimb ischemia, compared to non-implanted
KO mice and WT mice.
|
|
Tumor angiogenesis
The growth curves for implanted B16-F1 melanoma
cells and QRsP-11 fibrosarcoma cells were much reduced
in KO mice compared to WT mice. Consistently, the
survival rate was much greater in the KO than the
WT mice.
Analysis of angiogenesis within and around the tissue
surrounding the tumor showed angiogenic evidence was
reduced in the KO mice compared to WT mice. VEGF-expressing
macrophage infiltration was reduced in the KO mice
compared to the WT mice in the tissue surrounding
tumors. Recent studies by other investigators have
shown that tumor-associated macrophages are released
by angiogenic cytokines including VEGF. These cytokines
can induce tumor-related angiogenesis.
|
|
Clinical implications
These investigators hypothesize that marked inhibition
of the AT1 receptor early after ischemia may have
an adverse effect on subsequent ischemic tissue injury
by inhibiting physiological angiogenesis.
The effects of the Ang II type 1 receptor blocker
on the incidence and mortality in cancer patients
warrants further clinical investigation.
An ARB alone did not improve prognosis after a myocardial
infarction (MI) in the VALIANT study. But, ACE inhibitor
therapy plus an ARB improved prognosis in patients
with heart failure in the VAL-HeFT and CHARM trials.
Therefore, in patients with an acute MI, especially
early after the infarction, the marked inhibition
of the AT1 receptor may reduce the subsequent angiogenic
response. Thus, these investigators believe that the
addition of an ARB to an ACE inhibitor may be recommended
at 4-6 months after the onset of infarction.
|
PAGE
TOP
|
Local
Renin-Angiotensin System Induces Ventricular Remodeling
through Oxidant Stress-mediated Breakdown of Extracellular
Matrix in Rats with Salt-Sensitive Hypertension
Yasuki Kihara
Kyoto University Graduate School
of Medicine, Kyoto, Japan
|
|
Although the renin-angiotensin system (RAS) may play
a major role in left ventricular (LV) remodeling,
the precise mechanisms are not fully characterized.
To address this question, these investigators conducted
experiments in Dahl salt-sensitive (DS) rats, a relevant
model for LV remodeling, and Dahl salt-resistant controls
(DR). The DR controls maintain their normotensive
state, normal LV function, and normal LV versus body
weight ratio. In contrast, the DS rats are hypertensive,
with significant concentric hypertrophy at age 11
weeks, normal LV function and a calculated LV wall
stress within the normal limit. Thus, the left ventricle
is well compensated against overload at 11 weeks.
But, at about 15-17 weeks, eccentric hypertrophy and
hypofunction of the LV is seen. The rats die of pulmonary
congestion very rapidly. Thus, this is a good model
to see the process from a compensated left ventricle
to LV remodeling or heart failure.
In the so-called low renin, high Angiotensin II
(Ang II) models, these investigators previously showed
that LV tissue angiotensinogen, ACE, and Ang II is
upregulated at the stage of LV hypertrophy.
They also showed in the DR and DS models that the
extracellular metalloproteinase (MMP) is markedly
activated during LV remodeling. MMP plays an important
role in the degradation of extracellular matrix. MMP
is at a normal level in both the DS and DR rats in
the LVH phase, but is markedly increased in the DS
rats after the transition to heart failure (from 0.9
at 11 weeks to 1.7 at 17 weeks; p<0.05). A tight,
linear relation between MMP activation and LV diameter
and LV systolic wall stress was found.
|
|
Study design
The goals of the present study were to 1) elucidate
the roles of the local RAS that may induce LV remodeling,
2) trace the signaling process between Ang II and
MMP activation, and 3) test the hypothesis that a
direct inhibition of MMPs could block LV remodeling
in a manner independent of tissue RAS activation.
DS rats were fed an 8% high-salt diet after 6 weeks
of age, and after 11 weeks the DS rats were divided
into 4 groups for chronic pharmacologic interventions:
1) control group, given 0.5% CMC solvent twice daily,
2) ARB group, given telmisartan 5 mg/kg once daily,
3) MMPi group, given the MMP inhibitor ONO-4817 (100
mg/kg twice daily), and 4) combined ARB and MMPi group.
As a reference, the DR rats were fed the same diet.
Assessments were animal survival for each group,
serial in-vivo echocardiographic study to quantify
LV size and function, electron microscopic study to
visualize the extracellular matrix (ECM) degradation,
and immunohistochemical staining to determine the
LV tissue damage by oxidant stress. Quantitative,
real-time PCR and Western blotting were performed
to measure the activation of oxidant stress signaling.
|
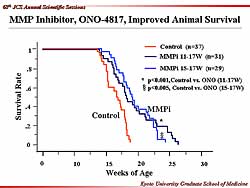 |
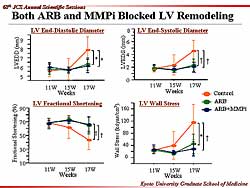
| Figure
1. Animal survival was improved with the MMP inhibitor
ONO-4817. |
| Click
to enlarge |
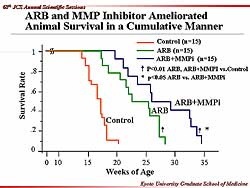 |
| Figure
2. Animal survival was ameliorated with an angiotensin
receptor blocker and the MMP inhibitor. |
| Click
to enlarge |
|
Improved survival with the MMP inhibitor was found
in the animals with heart failure. The animals
in the control group died quire rapidly, by 20 weeks
of age, while the MMP inhibitor initiated at 11 weeks
extended survival to about 25 weeks in the heart failure
animals (Figure
1). Animal survival was also about 25 weeks when
the MMP inhibitor was initiated at 15 weeks. Thus,
the MMP inhibitor acts along with the activation of
MMP at the heart failure transition phase.
A substantial improvement in survival to nearly
30 weeks was also seen with the ARB, with no reduction
in blood pressure. Importantly, combined ARB and MMPi
significantly improved survival to about 35 weeks
(Figure
2). This suggests that the ARB and MMPi could
be working via different cellular actions, and worked
synergistically to improve survival.
Figure
3 summarizes the echocardiographic study. In the
control animals, the LV diameter and LV end systolic
diameter began to increase at 15 weeks of age and
continued to 17 weeks, whereas the increase in LV
wall stress began at 11 weeks, and there was a decrease
in LV fractional shortening. In the ARB group and
the ARB plus MMPi group there was a similar trend
from 11 weeks in LV diameter, LV end systolic diameter,
LV wall stress, and fractional shortening. These 2
groups maintained their LV shape and function at normal
levels.
In the LVH stage, there was a very dense network
between the cardiomyocytes on electron photomicrography.
Surprisingly, the ECM became very thin after the heart
failure transition. The ECM degradation was lessened
with the ARB, and combined ARB plus MMPi maintained
ECM.
Marked activation of oxidative stress was seen during
heart failure transition, while none was seen in the
LVH stage, based on findings from the immunofluorescent
staining with HNE (4-hydroxy-2-nonenal), the fingerprint
of oxidant stress on membrane proteins, and with 8-OhdG
(8-hydroxy-2’-deoxyduanosine), the fingerprint
of oxidant stress on nuclear proteins. Notably, the
ARB markedly reduced the oxidative stress, but the
MMPi did not block oxidative stress.
NAD(P)H could be the critical mediator of the oxidative
stress in the presence of Ang II activation. Measurement
of NAD(P)H oxidized activation in mRNA and Western
blotting showed that the protein levels of P47phox
were substantially increased in heart failure compared
to LVH, but in the presence of the ARB, the P47phox
level was normal. But, the activation of P47phox
was not blocked by the MMPi. Thus, the subcellular
mechanism for the ARB and the MMPi are completely
different.
|
|
Summary
In an animal model of the process from ventricular
remodeling to heart failure, these investigators showed
that 1) chronic administration of an ARB and a MMP
inhibitor improved animal survival, LV shape and function,
2) this improvement was associated with preservation
of ECM, 3) tissue Ang II affected ECM degradation
through activation of NAD(P)H oxidase-mediated oxidant
stress, and 4) tissue MMP activation directly caused
ECM degradation independent of tissue Ang II activation.
Tissue Ang II activation causes ECM degradation
and plays critical roles in the process of ventricular
remodeling. ARBs, such as telmisartan, effectively
suppress this detrimental process. Exogenous administration
of MMP Inhibitors, such as ONO-4817, may serve as
an adjuvant therapy to further suppress the process
of ventricular remodeling in combination with the
angiotensin blockade.
|
PAGE
TOP
|
Effect
of Eplerenone, a Novel Selective Aldosterone Receptor
Antagonist in Salt-Sensitive Hypertension
Yoshiyu Takeda
Kanazawa University, Kanazawa,
Japan
|
|
The reported deleterious effects of aldosterone include
myocardial fibrosis, vascular injury and inflammation,
endothelial dysfunction, and progressive renal injury.
Although the RALES trial showed a 30% risk reduction
for death in heart failure patients, the side effects
profile of spironolactone is problematic. The specific
aldosterone receptor antagonist eplerenone reduced
cardiovascular death by 21% in post-AMI heart failure
patients.
About 50-60% of Japanese patients with hypertension
are salt-sensitive. The Dahl salt-sensitive rat (DS)
is a good model for salt-sensitive hypertension and
heart failure. This group previously reported that
in DS rats, a high sodium diet increased blood pressure,
reduced plasma renin activity, reduced plasma aldosterone,
and reduced vascular 11ß-HSD11 activity and
mRNA expression. The expression of mineralocorticoid
receptor RNA was increased by a high sodium diet.
Therefore, these investigators hypothesized that a
local excess of aldosterone because of reduced 11ß-HSD11
activity is a major cause of salt-sensitive hypertension
in rats.
|
|
Study design
This study sought to clarify the mechanisms responsible
for the anti-hypertensive and anti-hypertrophic effects
of eplerenone in salt-sensitive hypertension.
DS rats (n=20) were treated with a low salt diet,
high salt diet or high salt diet plus eplerenone (100mg/kg/day)
for 12 weeks. Blood pressure, plasma renin activity
(PRA), plasma aldosterone, heart weight, the expression
of mRNA of type I angiotensin receptor (AT1R), angiotensinogen,
angiotensin converting enzyme (ACE) and endothelial
nitric oxide synthase (eNOS) were measured. Real-time
PCR methods were used to quantify the mRNA of each
gene.
|
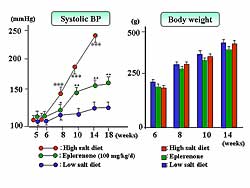 |
| Figure
1. Eplerenone prevented the increase in systolic
blood pressure and body weight caused by the high-salt
diet in the Dahl salt-sensitive rats, while the
body weight was similar in both groups.
|
| Click
to enlarge |
 |
| Figure
2. A high sodium diet decreased plasma renin activity
and aldosterone levels. PRA activity was increased
slightly by eplerenone, which had no effect on
aldosterone. |
| Click
to enlarge |
 |
| Figure
3. Eplerenone improved survival, while a high-salt
diet worsened survival. |
| Click
to enlarge |
|
In DS rats, a high-salt diet increased blood pressure
to about 250 mm Hg, but eplerenone treatment prevented
the increase (160 mm Hg). Body weight was similar
in each group (Figure
1).
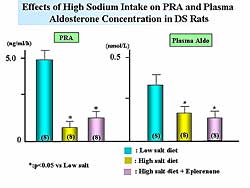
The high sodium diet significantly decreased the
PRA and plasma aldosterone levels. Treatment with
eplerenone slightly increased PRA but did not affect
the aldosterone concentration (Figure
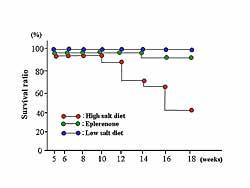
2). As illustrated in Figure
3, eplerenone improved the survival ratio.
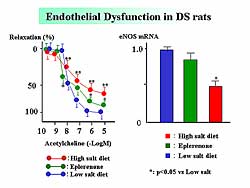
Acetylcholine-induced relaxation was reduced by
a high sodium diet, but treatment with eplerenone
partially improved this relaxation (Figure
4). Aortic eNOS mRNA expression was reduced by
a high salt diet, but this was attenuated with eplerenone.
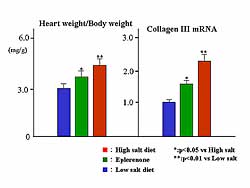
The heart weight/body weight ratio and collagen II
mRNA were increased by a high sodium diet, while eplerenone
treatment improved the heart weight and cardiac fibrosis
(Figure
5).
Cardiac calcineurin mRNA and cardiac Angiotensin
type 1 receptor mRNA, cardiac ACE mRNA, and cardiac
Ang mRNA were increased by a high sodium diet, and
were improved by eplerenone (Figure
6). So, salt-sensitive hypertension is a type
of local aldosterone excess state, and the specific
aldosterone receptor blocker eplerenone improved hypertension
and cardiac hypertrophy and fibrosis (Figure
7).
|
 |
| Figure
4. The effect of eplerenone, high salt diet, and
low salt diet on endothelial dysfunction in rats.
|
| Click
to enlarge |
|
 |
| Figure
5. The effects on heart weight/body weight ratio
and collagen III mRNA by eplerenone, high salt
diet, and low salt diet. |
| Click
to enlarge |
|
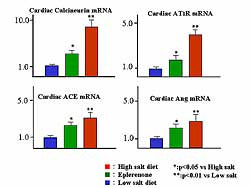 |
| Figure
6. The effects on cardiac calcineurin mRNA, cardiac
AT1R mRNA, cardiac ACE mRNA, and cardiac Angiotensin
mRNA. |
| Click
to enlarge |
|
|
PAGE
TOP
|
Blockade
of Cardiac Aldosterone Production as a New Therapeutic
Strategy of Heart Failure
Michiro Yoshimura
Kumamoto University, Kumamoto,
Japan
|
|
Production of aldosterone in the human heart
Recent evidence showed that aldosterone blockade
using either spironolactone or eplerenone improved
the prognosis of patients with heart failure. Based
on this evidence, it can be speculated that there
are unknown effects of aldosterone and that there
is the possibility of extra-adrenal synthesis of aldosterone.
This group previously reported that BNP is secreted
from the ventricles and that ACE is activated in the
ventricles of patients with heart failure. Aldosterone
is also secreted from the ventricles of the patients
with heart failure, but not from the ventricles of
control subjects. Another experiment confirmed the
expression of the CYP11B2 gene and aldosterone production
in the human heart; in autopsied human hearts expression
of the CYP11B2 gene was higher than in patients who
died of cancer without heart failure.
|
|
Aldosterone actions currently proved
Several reports have shown that aldosterone induces
inflammation in many organs including the heart and
blood vessels. This group hypothesized that aldosterone
has many actions, including ACE gene expression, creating
a circular cascade within the cardiac renin-angiotensin-aldosterone
system (RAAS).
In experiments in cultured rat neonatal cardiomyocytes
using real-time PCR, this group showed that aldosterone
increases ACE gene expression, which was completely
suppressed by spironolactone, a mineralocorticoid
receptor antagonist. This result confirms the presence
of the circular cascade with cardiac RAAS via the
mineralocorticoid receptor. Interestingly, they found
this action is cell-specific and is species different.
Nevertheless, this action can be seen in the left
ventricle of patients with heart failure, based on
preliminary data by this group.
|
|
Inhibitory effect of natriuretic peptides
on cardiac aldosterone production
The natriuretic peptide family comprises ANP, BNP,
and CNP, which have many important actions. It is
well known that ANP and BNP secrete from the failing
heart
Angiotensin II did not increase CYP11B2 gene expression
in cultured rat neonatal cardiomyocytes. However,
pre-treatment of the cells with HS1421, a GC-A receptor
antagonist suppressing endogenous effects of natriuretic
peptide, Angiotensin II significantly increased the
CYP11B2 expression. This results confirms the inhibitory
action of natriuretic peptide on cardiac aldosterone
production and suggests that if there were no natriuretic
peptides in the failing heart, it could produce considerable
aldosterone.
|
|
Production of dehydroepiandrosterone (DHEA)
in the human heart
Considering the presence of the cascade to produce
aldosterone, they hypothesized this cascade could
produce CYP17 in the human heart. Real-time PCR of
the enzymes required for steroid synthesis of human
hearts obtained at autopsy confirmed the presence
of CYP17. Then, during cardiac catheterization they
performed direct sampling of DHEA and aldosterone
in patients with heart failure and control subjects.
They showed that DHEA is secreted from the heart of
control subjects, but not from the heart of patients
with heart failure. In contrast, aldosterone is secreted
from the heart of patients with heart failure, but
not from the heart of control subjects. The DHEA/aldosterone
ratio showed that the value was significantly lower
in patients with heart failure compared to the control
subjects, especially in the coronary sinus.
Aldosterone is thought to be a hormone for oxidation
and inflammation, whereas DHEA is thought to a hormone
for anti-oxidation and anti-inflammation. Thus, they
examined the possible inhibitory action of low-dose
DHEA on cardiac hypertrophy induced by ET-1 in cultured
rat cardiomyocytes. Pre-treatment with DHEA suppressed
cardiac hypertrophy induced by ET-1, both for cell
size and BNP gene expression. So, even though the
receptor for DHEA has not been elucidated, it appears
that DHEA plays an important role in cardiac protection.
|
|
Production of adrenocorticotropic hormone
(ACTH)
Based on the previous evidence, they thought that
ACTH may be also secreted from the human heart. During
cardiac catheterization, direct sampling was performed
in patients with hypertension and in control subjects.
ACTH was secreted from the ventricles of patients
with hypertension, but not from the ventricles of
the control subjects.
They found a close relationship between cardiac
ACTH and cardiac aldosterone. This result implies
that ACTH produced in the ventricle continuously induces
aldosterone synthesis in the ventricle. It is interesting
to see this action of ACTH, because it is thought
to suppress aldosterone synthesis in the adrenal gland
in the chronic phase, thus the action of ACTH would
be in both the heart and the adrenal gland. They hypothesize
that ACTH and Angiotensin II collectively induces
aldosterone synthesis in the heart.
Aldosterone is stimulated by Angiotensin II in the
failing heart. ACTH produced in the heart stimulates
aldosterone synthesis in the failing heart. Natriuretic
peptides suppress aldosterone synthesis. Interestingly,
there is an unstable balance between aldosterone and
DHEA. They have studied only part of the cascade of
RAAS, ACTH, and steroids in the human heart. To understand
this complex cascade, the many possible relationships
among these must be investigated.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2004
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|