|
|
|
|
| Genome Informatics for the Cardiovascular
System |
|
|
|
|
|
DNA
Microarray Analysis of Genes in Human Atrial Myocardium
Keiji Yamamoto
Jichi Medical School, Tochigi,
Japan
|
|
Molecular studies in patients are needed to develop
novel treatment strategies. DNA microarray analysis
is a powerful technology that offers a comprehensive
profile of gene expression. Work performed by this
group illustrates the powerful benefits of genome
informatics.
|
|
Study of human right atrium
|
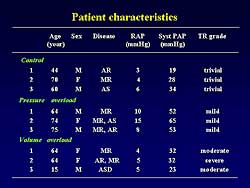 |
| Figure
1. The patient characteristics in the three study
groups. |
| Click
to enlarge |
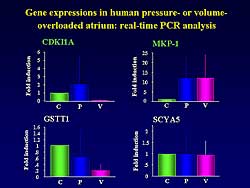 |
| Figure
2. The gene expression in human pressure- or volume-overload.
|
| Click
to enlarge |
|
In 9 patients undergoing cardiac surgery, most with
valvular heart disease, this group investigated the
transcriptional profile of genes induced in pressure-overload
or volume-overload human atria.
The patients were divided into 3 groups based on
pressure data and echocardiographic findings: 1) control,
2) pressure-overload (mean right atrial pressure >
7 mm Hg), 3) volume overload (moderate or severe tricuspid
regurgitation). The patient characteristics are shown
in Figure
1. The expression of 2,139 well-characterized
functional genes, such as cell cycle protein, growth
factors, and cytokines was investigated.
On DNA microarray analysis, only 4 genes were expressed:
cyclin-dependent kinase inhibitor 1A (CDKI1A), MAP
kinase phosphatase-1 (MKP-1), glutathione S-transferase
theta 1 (GSTT1), and small inducible cytokine A5 (SCYA5)
(Figure
2).
In the pressure overload group, CDKI1A and MKP-1
were significantly increased, compared to the control
and volume overload groups (12-fold induction versus
< 4-fold in control and volume overload for CDKI1A,
and for MKP-1 a 25-fold versus < 10-fold induction
in control and volume overload). In contrast, the
expression of GSTT1 and SCYA5 was the same in each
group.
The increased expression of CDKI1A and MKP-1 mRNA
in the volume overload group was confirmed by real-time
PCR. For this analysis, primers of the 4 expressed
genes were designed for real-time PCR. To confirm
the preparation expressions of selected genes in the
pressure overload group, cDNA from the same RNA used
in the microarray analysis was subjected to real-time
PCR.
|
|
Analysis of MKP-1 and CDKI1A
|
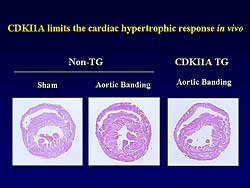 |
| Figure
3. CDKI1A limited cardiac hypertrophy response
in vitro. |
| Click
to enlarge |
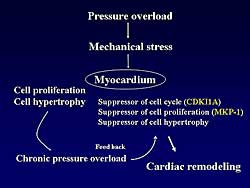 |
| Figure
4. The effect of pressure overload on mechanical
stress and the myocardium, leading to cardiac
remodeling. |
| Click
to enlarge |
|
MKP-1 has dual catalytic activity toward phosphotyrosine-
and phosphothreonine-containing proteins, and is known
to inactivate extracellular signaling-regulated protein
kinases, possibly JNKs and p38-MAP kinases. CDKI1A
functions by binding to and inactivating a number
of the cyclin and cyclin-dependent kinase complexes,
including cyclin-dependent kinase 2. CDKI1A is a regulator
of the G1-S cell cycle checkpoint. In the adult heart,
the role of the cell cycle, including CDKI1A, is unknown.
Mechanical stress induced MKP-1 and CDKI1A protein
expression in cultured neonatal rat cardiomyoctyes
in their experiments.
Bueno and colleagues had demonstrated a dramatic
attenuation of cardiac hypertrophy in response to
aortic banding in alpha-myosin heavy chain cardiac
specific MKP-1 expressing transgenic mice. Therefore,
these investigators made transgenic mice expressing
CDKI1A under the control of an alpha-myosin heavy
chain cardiac specific promoter, in collaboration
with investigators at Tokyo University. In wild-type
mice, abdominal aortic banding induced left ventricular
hypertrophy (LVH), while in the CDKI1A transgenic
mice the LV was only dilated a little after aortic
banding (Figure
3). In the CDKI1A transgenic mice, attenuation
of LVH development was also found. These findings
suggest the importance of CDKI1A as a counter-balancing
regulatory factor in the heart. The precise role of
CDKI1A in cardiac hypertrophy is the subject of further
study by this group.
Based on the present findings, these investigators
speculate that a suppressor of cell cycle, proliferation,
or cell hypertrophy may play a critical role in the
pathophysiology of pressure overload (Figure
4).
|
|
Molecular mechanisms in AF
|
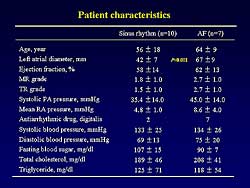 |
| Figure
5. The patient characteristics in the analysis
of atrial fibrillation. |
| Click
to enlarge |
 |
| Figure
6. Thirty-three genes specific for atrial fibrillation
were found. |
| Click
to enlarge |
|
Gene expression profiling of human atrial myocardium
with AF using DNA microarray analysis was performed
by this group.
A comprehensive analysis of the regulatory mechanisms
involved in AF had not been performed previously,
and molecular research has mainly focused on ion channels
and proteins involved in calcium handling.
In 17 fresh cardiac specimens (10 sinus rhythm (SR),
7 AF) from the right atrial appendage, the expression
profile of 12,000 human genes was investigated with
mRNA from the atria using Human Genome U95A arrays
(Figure 5). The
left atrial diameter was significantly greater in
the AF group than in the SR group (67 mm vs 42.7 mm;
p<0.001). However, there were no other differences
between the 2 groups.
The DNA microarray analysis identified 33 AF-specific
genes that are activated compared to SR (Figure
6). Of these, macrophage migratory inhibitory
factor and NF-IL6-beta are involved in inflammation.
In contrast, they found 63 SR-specific genes. Sarcoplasmic
reticulum Ca2+-ATPase 2 is a calcium ion regulatory
protein and Connexin 43 is one of the Gap junctions
that are clusters of closely packed channels.
NF-IL6-beta mRNA induction in the AF group was confirmed
by real-time PCR. A novel and inflammatory mechanism
may promote the initiation and persistence of AF,
potentially by inducing structural and electrical
remodeling of the atria.
Connexin 43 and SERCA2 expression in the AF group
was significantly lower than that in the SR group.
The abnormalities in inter-cellular calcium ion handling
and changes of the atria in chronic AF patients may
be involved in the initiation and perpetuation of
AF. Changes of Connexin 43 expression may affect conduction
velocity and induce and sustain persisting AF.
|
|
Summary
Novel and inflammatory mechanisms may be involved
in the mechanism of AF.Decreased SERCA2 and connexin
43 mRNA expressions may contribute to the molecular
mechanism of AF. Although the roles of many genes
including ESTs in the heart remain unknown, the genes
screened in this study may provide insights into the
initiation or perpetuation of AF and the pathophysiology
of atrial remodeling.
|
PAGE
TOP
|
Systematic
Linkage between the Genomic and Clinical Database
of Cardiovascular Disease to Find New Pathophysiology
or Effective Treatment
Masafumi Kitakaze
National Cardiovascular Center,
Osaka, Japan
|
|
This presentation illustrated the use of 4 different
strategies to elucidate candidate genes for chronic
heart failure (CHF). This group states that these
strategies, which use genomics or pathogenomic sciences,
combined with molecular biology, gene expression,
physiology, and clinical medicine, will identify the
genes for certain cardiovascular diseases, and lead
to novel therapies.
The 4 strategies used to identify the candidate
gene for CHF amongst the 35,000 available genes, were:
1) knowledge from the existing literature, 2) identify
common genes that are upregulated or downregulated
in the failing myocardia in the studied species, 3)
identify heart failure-related genes by linking clinical
parameters to gene expression level, and 4) identify
genes modulated by drugs known to be cardioprotective,
such as ACE inhibitors.
DNA chip analysis was performed in failing myocardium
samples obtained from 1) patients with CHF (n=20),
2) canine failing myocardium produced by rapid pacing
(n=12), and 3) murine failing myocardium produced
by aortic banding (n=60).
|
|
Extending literature-based knowledge to pathophysiology
of CHF
In the 20 samples of human failing myocardium, of
the 10,000 known genes, the expression of about 3%
were significantly modulated, that is, about 300 genes
were upregulated or downregulated.
The adenosine-related genes among the modulated
genes in the failing myocardium were examined, because
adenosine is known to be cardioprotective against
ischemia/reperfusion injury. The adenosine A2a and
A3 receptors are downregulated in the failing myocardium,
compared with a commercially-available control myocardium,
on DNA microarray analysis.
To prove their hypothesis that the downregulation
of the adenosine receptor is linked to the pathophysiology
of CHF, they administered the adenosine agonist 2-chloroadenosine
in the murine heart failure model produced by thoracic
aortic banding (TAC). TAC produced cardiac hypertrophy
and 2-chloroadenosine potently inhibited cardiac hypertrophy.
The TAC-increased lung edema was strongly attenuated
by the adenosine analog.
Therefore, they conclude that the adenosine A2a
receptor is downregulated in patients with CHF and
the restoration of the adenosine receptor activation
improves heart failure in the mouse model.
A difference in the SNP of the A2a receptors was
found between the control and patients with dilated
cardiomyopathy (DCM). This strengthens the notion
that the decreases in the effect of adenosine following
a genetic abnormality of the adenosine receptors may
be one of the mechanisms that progresses CHF.
To test the effect of adenosine in the clinical
setting, they administered the adenosine potentiator
dipyradimole in patients with CHF. A 6-month administration
of dipyradimole improved NYHA functional class, and
increased the ejection fraction (EF) and exercise
capacity. These results suggest that augmentation
of the effect of adenosine improves CHF.
|
|
Identifying commonly-modulated genes
|
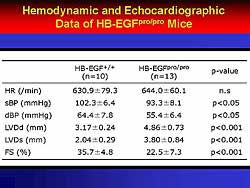 |
| Figure
1. The hemodynamic and echocardiographic data
of HB-EGFpro/pro mice. |
| Click
to enlarge |
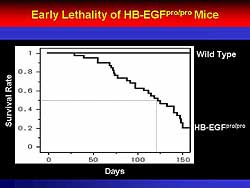 |
| Figure
2. The HB-EGFpro/pro mice died early compared
to wild-type mice. |
| Click
to enlarge |
|
The second strategy was to find commonly-modulated
genes in the human, canine, and murine failing myocardium
using DNA microarray, and test whether such genes
are responsible for heart failure.
In the human heart, the modulated genes were linked
to clinical parameters, such as BNP, LVEF, and pulmonary
arterial pressure. Of these, approximately 80 genes
are only modulated in the human failing myocardium,
including heparin-binding epidermal growth factor
(HB-EGF). HB-EGF is upregulated in the failing canine
myocardium. In the murine model, HB-EGF was closely
correlated to changes in DNA expression levels.
HB-EGF is closely related to the pathophysiology
of HF, based on experiments by this group. In knockout
mice, the deletion of HB-EGF function did not cause
systemic blood pressure changes, but provoked enlargement
of ventricular diameter and decreased contractile
function (Figure
1). Further, the knockout mice, but not the wild
type mice, died of cardiovascular (CV) death at about
150 days (Figure
2).
Further investigation also suggested that abnormalities
of HB-EGF genes may contribute to the pathophysiology
of the human failing heart. Examination of the differences
in SNPs related to HB-EGF and ADAM-12, which shares
membrane-bound HB-EGF, between control subjects and
patients with DCM revealed that the SNPs at exon 3
and exon 5 were found in patients with DCM.
|
|
Linkage analysis between gene expression and
clinical parameters
|
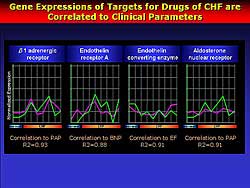 |
| Figure
3. The expression of genes that are targets for
heart failure drugs and their correlation to clinical
parameters. |
| Click
to enlarge |
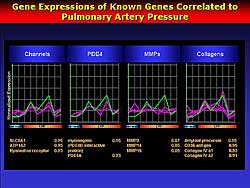 |
| Figure
4. The expression of genes known to be correlated
to pulmonary artery pressure. |
| Click
to enlarge |
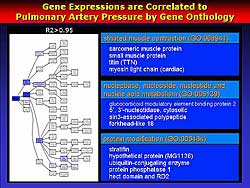 |
| Figure
5. The correlation between gene expression and
pulmonary artery pressure by gene onthology. |
| Click
to enlarge |
|
The correlation between the change in gene expression
and plasma BNP levels, EF or pulmonary arterial (PA)
pressure was examined. They found 212 genes tightly
and positively linked to PA pressure, and 15 genes
were inversely linked to changes in PA pressure.
Next, they investigated if there was a relation
between the genes related to drugs used to treat CHF
and the genes related to PA pressure. The beta-1 adrenergic
receptor, endothelin receptor A, endothelin converting
enzyme, and aldosterone receptor are tightly linked
to CHF, and this analysis showed these genes are also
well-correlated to the PA pressure changes (Figure
3). Therefore, they believe this strategy works
for mining for novel genes for heart failure.
They further classified the CHF-related genes according
to the cellular function. Genes related to channels,
phosphodiesterase, matrix metalloproteinase, and collagen
were tightly linked to the PA pressure (Figure
4). This suggests that networks of these gene
changes contribute to the pathophysiology of heart
failure.
The PA pressure-related genes were examined based
on gene onthology, and showed that many unexpected
genes are in the groups of striated muscle contraction,
nucleobase, nucleoside, and nucleotide, nucleic acid
metabolism, and protein modification (Figure
5). These genes are tightly related to PA pressure.
The same genes are modulated when linking was performed
to ejection fraction. Among these genes, a second
screening was performed to determine the relation
to cellular survival or death. They transfected the
candidate genes for CHF to the rat cardiomyocyte and
examined the amino acid uptake and cell survival in
this system. The X2 genes decreased amino acid uptake,
and X5 genes are increased in cell survival. This
method verified that X3 and X5 genes are crucial for
cell survival. Knockout mice with these genes are
being developed for further study.
Haplotype analysis revealed that the SNP pattern
of 9 genes is significantly different. These genes
are tightly related to CHF and testing these genes
is worthwhile to determine whether they are related
to the pathophysiology of CHF.
|
|
Identify genes modulated by drugs
In myocardial samples from failing canine myocardium,
of the 20,000 genes examined by DNA microarray, 2,005
genes had active significant signals as assessed by
Cy3 or Cy5. Among these genes, 50 genes were upregulated
and 25 genes were downregulated—and several of
these were potently modulated by the ACE inhibitor
temocapril. ACE inhibitors are known to be cardioprotective.
Further testing of these genes is worthwhile.
HB-EGF was found to be upregulated by ACE inhibitors
and ARBs compared to the failing myocardium without
any drugs. These results also suggest that HB-EGF
plays a role in the pathophysiology of HF.
|
PAGE
TOP
|
Establishment
of Clinical Data Management System and Its Practical
Application to Genetic Epidemiology in Cardiovascular
Medicine
Tsutomu Yamazaki
University of Tokyo, Tokyo, Japan
|
|
The trinity of genome information, clinical information,
and information technology is indispensable to research
in the field of genome informatics in cardiovascular
disease (CV). Multivariate analysis using genetic
and environmental factors is necessary to investigate
the mechanisms of CV disease. The gene-environment
interaction also influences the susceptibility of
the genesis of CV disease. Thus, a combination approach
to genetic and clinical factors has been used by these
investigators.
|
|
Database development
Comprehensive clinical information from about 3,000
patients admitted to the University of Tokyo has been
collected in their clinical database. DNA samples
from 1,300 patients who provided signed informed consent
are also available for SNP analysis. The genetic study
conducted by this group was approved by the Ethics
Committee of the University of Tokyo.
More than 500 clinical parameters, such as basic
characteristics, diagnosis, risk factors, noninvasive
examination, coronary angiography, percutaneous intervention,
bypass surgery, cardiac events, complications, and
medications, are stored in the clinical database.
Analysis of about 50 SNPS known to be implicated in
the pathogenesis of CV disease was performed, such
as ACE, angiotensinogen, and angiotensin II type 1
receptor.
|
 |
| Figure
2. The chronological data in one patient allows
for accessing more detailed information by just
clicking on an item of interest. |
| Click
to enlarge |
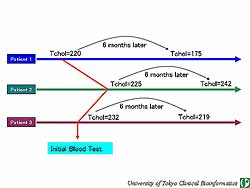 |
| Figure
3. Side-by-side analysis, such as this analysis
of changes in cholesterol levels, can be used
for epidemiologic studies. |
| Click
to enlarge |
 |
| Figure
4. Chronological view of the QCA data in one patient
in the database. |
| Click
to enlarge |
|
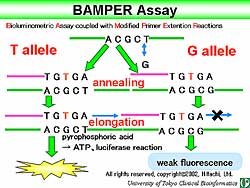
The Bioluminometric Assay coupled with Modified Primer
Extension Reactions (BAMPER) assay was introduced
by this group of investigators for analyzing human
genetic variations (Figure
1). After PCR, probes are added and pyrophosporic
acid is produced, which is converted to ATP, to analyze
the luciferase reaction system.
Figure
2 illustrates the chronological sequence of the
clinical information in one patient, such as clinical
symptoms, hospitalization, complications, noninvasive
testing, intervention, and prescriptions. Detailed
data on any item can be accessed easily by clicking
on any item of interest. Variations in total cholesterol
can be investigated by arranging the levels in a side-by-side
manner (Figure
3). This analytical method provides for easily
conducting epidemiological studies, including prospective
and transversal studies.
Medical treatment records of patients at the University
of Tokyo are maintained in an electronic database
system, which provides the ability to perform chronological
reference and prospective and transversal studies
and analysis of medical treatment data mainly through
data mining.
Data from coronary artery quantitative analysis
are included in the database. The QCA data is automatically
transferred to the database. Figure
4 illustrates a chronological view of the QCA
data in one patient, providing an easy overview of
the patient status, including the progression or regression
of disease in a vessel.
For a transversal study, the patients in the database
were divided into 2 groups based on the presence or
absence of diabetes. The ratio of patients who developed
restenosis can be easily determined and these findings
can be automatically shown in real-time (Figure
5).
Data mining is an exhaustive method that is generally
accepted and used in recent years.
An efficient search of a large database can be easily
performed using specified criteria.
The table in Figure
6 illustrates the use of data mining. About 81%
of the patients with the specified premise had a myocardial
infarction (MI). Support means the ratio of patients
who have both the premise and conclusion. The odds
ratio shows the patients who have the premise and
are susceptible to MI, about 4.7 times, compared to
the patients who don’t have this premise.
|
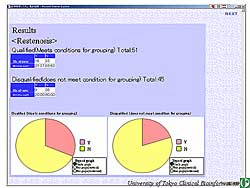 |
| Figure
5. Restenosis in relation to the presence or absence
of diabetes can be easily determined using the
database. |
| Click
to enlarge |
|
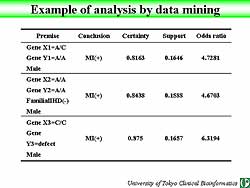 |
| Figure
6. An example of real-time data mining based on
specified criteria. |
| Click
to enlarge |
|
|
Examples from their database system
|
 |
| Figure
7. Logistic regression analysis performed in patients
with CAD and the Met allele. |
| Click
to enlarge |
 |
| Figure
8. Genetic polymorphisms implicated in insulin
resistance had no association with the severity
of CAD |
| Click
to enlarge |
|
Cholesterol esters are transferred via the ABCA1
protein and bind to the lipid-poor Apo-A1 particles.
Therefore, a variation in the ABCA1 gene, which regulates
the cholesterol influx from the cell, may modify the
protein function leading to the alteration of the
lipid profiles, such as HDL cholesterol and Apo-A1
levels. So, this group investigated whether a common
variant of the ABCA1 gene at the position of Ile823Met,
may influence the plasma lipid profiles, including
HDL-C, and the susceptibility to coronary artery disease
(CAD) in the Japanese population in their database.
In 292 patients untreated for hyperlipidemia, they
found significantly higher HDL-C levels in patients
with Met/Met (49.4 mg/dl, p<0.05 vs Ile/Ile patients)
compared to patients with Ile/Ile (45.7 mg/dl) and
Ile/Met (49.8 mg/dl).
The correlation between the Met allele and CAD was
then analyzed. Distribution of the Met allele was
about 31% and 42% with or without CAD, respectively.
This suggests that the Met allele is associated with
significantly higher levels of HDL-C, resulting in
the inhibition of CAD. On logistic regression analysis,
age >65 years, hypertension, diabetes, hyperlipidemia,
and ABCA1 gene polymorphism (odds ratio 1.54) were
independent risk factors for CAD (Figure
7).
Adiponectin was recently shown to have anti-atherogenic
and anti-diabetic effects. Adiponectin, structurally
regulated by collagen, has been identified as a novel
adipose-specific gene product. The polymorphism at
position 276 of adiponectin was analyzed by this group
using ELISA. The G allele at position 276, thought
to be associated with diabetes or insulin resistance,
was conversely and linearly related to plasma adiponectin
levels in the Japanese patients in the clinical database.
Next, the severity of CAD and adiponectin polymorphism
was evaluated. The G allele at position 276 was significantly
associated with the severity of CAD only in obese
patients, but not in non-obese patients. However,
other genetic polymorphisms implicated in insulin
resistance, such as IRS1, IRS2, PPAR-gamma, and the
leptin receptor, had no association with the severity
of CAD (Figure
8).
|
|
Conclusion
The original database system established at this
institution supports genetic epidemiologic studies
and clinical investigation, including prospective
cohort study, clinical trials, and medical research,
especially on the adverse reactions of new drugs.
This system has great potential to elucidate the susceptibility
for CAD and previously unknown information, which
should contribute to personalized medicine and novel
therapeutics and new drug design for CV disease.
|
PAGE
TOP
|
Pharmacogenomics
and Pharamacoinformatics
Toshio Tanaka
Mie University School of Medicine,
Tsu, Japan
|
|
The goals of pharmacogenomics are to identify gene
clusters involved in determining the responsiveness
to a given drug, to distinguish responders and nonresponders,
and to predict side effects. This presentation focused
on drugability and drugable genomics.
The principle of molecular pharmacology is reductionist,
whereas the principle of pharmacogenomics is comprehensive
and individualized. Studying drugability using molecular
pharmacology provides information on the interaction
between medicine and the drug target, and some information
on disease associated genes. However, there is a large
gap between the therapeutic gene cluster and disease-gene
clusters.
|
|
Studies of cerebrovascular spasm and subarachnoid
hemmorhage
Drugability can now be researched using pharmacogenomics.
Therapeutic gene clusters, disease gene clusters,
and the interaction between these clusters can be
exploited to find potential therapeutic actions.
This group constructed an original pharmacogenomic
database, in which drug action or disease information
can be translated into gene clusters. This enables
comparing the relation between therapeutics and disease
in the common genomic database. The database comprises
3 dictionaries: gene, chemical, and disease.
To study cerebral vasospasm, this group used their
pharmacogenomic database. Cerebral vasospasm can be
defined as a delayed onset narrowing of the cerebral
artery, which can occur after subarachnoid hemorrhage
(SAH). The pathophysiology of cerebral vasospasm remains
unknown.
Transcription analysis of the differences in gene
expression on H-E staining of the basilar artery in
their rat model revealed that encoding for heme oxygenase
1 is highly upregulated by basilar artery by delayed
cerebral vasospasm.
Three genes (NOS1, ICAM1, EDM3) are related to both
HO1 and cerebrovascular disorders. Thus, they tried
to determine the mechanism of action of HO-1 in cerebrovascular
disorders. Antisense for HO-1 ODN in vivo inhibited
HO-1 induction in basilar artery and in deteriorated
cerebral vasospasm. This phenomenon was not observed
in the scrambled ODN. This data suggested that HO-1
has a potential protective function in cerebrovascular
spasm.
Administration of the potent HO-1 inducer hemin
significantly induced HO-1 and significantly improved
the diameter of the basilar artery by angiography.
This result suggests HO-1 induction is protective
of disease.
Other experiments by this group showed that the
low molecular weight (LMW) chemical compound AVS selectively
induced HO-1 in the basilar artery, only in the presence
of SAH. A single administration of AVS in vivo did
not induce the basilar artery. In their model, AVS
has a potential therapeutic action on cerebrovascular
spasm measured by angiography. AVS is being studied
in clinical trials, with one report of its efficacy
for cerebrovascular spasm in a small number of patients.
In other work by this group, they found that heat-shock
protein (HSP) 72 was greatly induced in SAH. Using
their database to check the relationship between HSP72-related
genes and cerebrovascular disease, they identified
1 gene related to HSP72 and cerebrovascular disorders.
To characterize the function and significance of HSP72
in cerebrovascular vasospasm, they used antisense
ODN, which is effective in suppressing HSP72 message
induction and inhibiting HSP72 induction at the protein
level. Antisense induction was protective of function
in this disease.
Oral GGA, another low molecular weight chemical,
dose-dependently induced HSP72 in the basilar artery.
GGA can induce HSP72 at the messenger level in the
presence of SAH. In the rat model, GGA administration
significantly improved cerebrovascular spasm, especially
day 1 to day 3.
|
PAGE
TOP
|
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2004
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|