|
|
||||||
|
|||||||
|
|||||||
|
|||
|
|||
Cardiovascular and Autonomic Actions of Leptins: Implications for the Cardiovascular Complications of ObesityAllyn L. MarkCarver College of Medicine
|
|

Leptin resistance and the loss of leptin action in cardiovascular disease was the focus of this lecture. Two concepts were reviewed: selective leptin resistance contributing to sympathetic overactivity and hypertension and obesity, and lipotoxicity of non-adipose tissues in the setting of loss of leptin action leading to insulin resistance and deficiency and potentially cardiac disease (Figure 1).
Cardiovascular actions of leptin and leptin resistance
Leptin is secreted from white adipocytes, where it circulates across the blood-brain barrier by an active transport mechanism to inhibit food intake and to increase thermogenic metabolism, thereby decreasing adiposity. There are rare Mendellian human counterparts of leptin deficiency, due to mutations in the leptin gene, and total leptin resistance, due to mutations in the leptin receptor.
But in most common human obesity, there are no mutations in either the leptin gene or the leptin receptor. Instead, there is evidence for partial leptin resistance, presumably secondary to abnormalities downstream signaling mechanisms from the leptin receptor, leading to compensatory hyperleptinemia. In addition, in obesity-induced hypertension, there is also evidence for sympathetic overactivity, which may contribute to the hypertension.
|
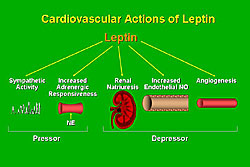
Leptin, like insulin, is now known to possess and array of cardiovascular (CV) and autonomous actions, including increases in sympathetic activity and adrenergic responsiveness, both of which could increase arterial pressure; increases in renal natriuresis, endothelial nitric oxide (NO), and angiogenesis, all of which could potentially contribute to decreases in arterial pressure (Figure 2).
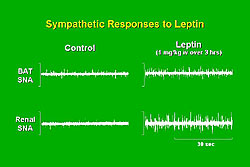
Marks and colleagues demonstrated in Sprague-Dawley rats that leptin increased sympathetic nerve activity in brown adipose tissue (Figure 3). Thermogenic metabolism in brown adipose tissue is known to be sympathetically mediated. Surprisingly, leptin also increased sympathetic activity in the kidney, which appears to be physiologically significant. For example, an elegant series of experiments by Ogawa and colleagues demonstrated that leptin produces sympathetically-mediated increases in arterial pressure. Transgenic mice overexpressing leptin from a liver promoter resulted in a 20-fold increase in leptin, and decreases in body weight and body fat that would be expected to lower arterial pressure. However, the increase in leptin was associated with significant increases in arterial pressure, which appeared to be sympathetically mediated, because they were accompanied by increases in urinary norepinephrine. The hypertension was acutely reversed by alpha-adrenergic blockade. So, the concept that leptin-induced increase in sympathetic activity contributes to increases in arterial pressure is supported by these and other studies of pharmacologic infusions of leptin.
Physiologic role of leptin regulation
To determine the physiologic role of leptin regulation of sympathetic activity and arterial pressure, Marks and colleagues measured arterial pressure directly in conscious wild-type and leptin-deficient obese ob/ob mice. The ob/ob mice had early onset severe obesity and complete leptin deficiency. Despite profound obesity, the leptin-deficient ob/ob mice had lower arterial pressure. Ogawa and colleagues have separately demonstrated that leptin reconstitution to physiologic levels restores the lower arterial pressure. Together, these studies support the concept that leptin produces physiologically significant sympathetically-mediated blood pressure actions.
|
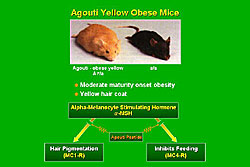

Agouti yellow obese mice have a moderate maturity onset of obesity and a yellow hair cat that are related to blockade of the actions of alpha-melanocyte stimulating hormone (alpha-MSH), which normally acts on melanocortin-1 receptors (MC1-R) in the terminal hair follicles to produce a dark haired coat, and on melanocortin-4 receptors (MC4-R) in the hypothalamus to inhibit feeding (Figure 4). The ubiquitous overexpression of Agouti peptide in the Agouti mice antagonizes the effects of alpha-MSH on the hair follicle, producing the yellow haired coat, and on the MC4-R, leading to increased food intake and decreased metabolism and obesity. Marks and colleagues found that the leptin-deficient ob/ob mice have profound obesity but lower mean arterial pressure (MAP; body weight 29 grams, MAP about 100 mmHg), but the Agouti mice have milder obesity yet have increases in arterial pressure (body weight 40 grams, MAP about 130 mmHg). Notably, the Agouti yellow obese mice were hypertensive, hyperleptinemic, and had leptin resistance unrelated to mutations in the leptin receptor. Ogawa and colleagues demonstrated that the elevated leptin contributes to the elevated arterial pressure.
Thus, the leptin-deficient ob/ob mice have lower arterial pressure, whereas the high-leptin Agouti mice have higher arterial pressure. So, the levels of leptin in obesity could be considered important in the blood pressure response to obesity. However, the Agouti mice are resistant to the metabolic effects of leptin (Figure 5). Hence, the paradox is: How does leptin contribute to blood pressure regulation in Agouti mice if the mice are resistant to the effects of leptin.
Selective leptin resistance
To address this paradox, Marks and colleagues proposed a concept of selective leptin resistance, analogous to the concept of selective insulin resistance in the metabolic syndrome. They speculated that in the Agouti mice, and perhaps other models of obesity, there is resistance to the effects of leptin on appetite and body weight, but perseveration of the effects of leptin on the sympathetic nervous system contributing to hypertension.
To test this concept, Marks and colleagues studied the effects of leptin on body weight, food intake, and renal sympathetic nerve activity in Agouti obese mice and in lean mice. As others have demonstrated, Marks and colleagues showed that leptin decreased food intake in the wild-type mice and this effect was attenuated in the Agouti mice. But, there was complete preservation of the renal sympathetic effects of leptin, indicating selective leptin resistance. They have found similar observations in an acquired diet-induced model of obesity in mice. These data suggest there is a selective leptin resistance phenomenon, with loss of appetite and body weight effects of leptin, contributing to obesity, leading to compensatory hyperleptinemia, which produces sympathetic activation and contributes to hypertension.
Lipotoxicity of non-adipose tissues
The Unger hypothesis, introduced by Roger Unger in 2000, states that the primary physiologic role of leptin is to prevent fatty acid accumulation in non-adipose tissues, for example, skeletal muscle, liver, pancreas, and heart.
Unger has demonstrated that leptin normally prevents lipotoxicity in non-adipose tissues, by acting on OB-receptors, leptin receptors, and cell membrane, activating PPAR-alpha and facilitating beta-oxidative metabolism to prevent lipid accumulation in non-adipose tissues. In the loss of leptin action due to leptin deficiency or leptin resistance, this mechanism is lost, leading to fatty acid accumulation in non-adipocytes, activating a toxic non-oxidative pathway of ceramide excess, iNOS activation, and apoptosis leading to cellular dysfunction. Unger has demonstrated that the consequences of this lipid accumulation in non-adipose tissue with loss of leptin action includes decreases in insulin action in skeletal muscle that is promoting leptin resistance, beta cell dysfunction leading to insulin deficiency, that is, the spectrum of type 2 diabetes; and interestingly, the accumulation of intracellular lipids in cardiac myocytes, leading to cardiac dysfunction, that can be reversed by interruption of the blockade of ceramide excess, iNOS activation, and blockade of apoptosis.
Congenital severe lipodystrophy
This syndrome is characterized in humans by the absence of adipocytes and severe insulin resistance and diabetes. This is a rare syndrome in which the loss of adipocytes also leads to severe refractory insulin resistance and diabetes. The leptin deficiency in this syndrome has been proposed to contribute to the insulin resistance. Oral and Garg demonstrated that leptin replacement therapy for lipodystrophy in refractory diabetes there were very high levels of triglycerides (about 8000 mg/dL), high levels of glycosolated hemoglobin (nearly 9%), and very low levels of leptin, despite aggressive treatment of the diabetes; progressive leptin therapy within the physiologic range resulted in a dramatic and sustained decrease in triglycerides and glycosolated hemoglobin, and a decrease in intracellular accumulation of lipids.
Contribution of Endoplasmic Reticulum-Initiated Apoptosis to Progression from Hypertensive to Failing HeartsTetsuo MinaminoOsaka University Graduate School of Medicine
|
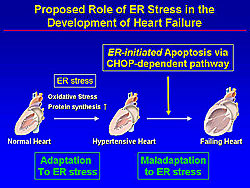
The failing heart is characterized by its enlargement and loss of cardiac myocytes. That endoplasmic reticulum (ER) plays a role in the failing human heart has been shown by the presence of areas of vacuolization consistent with GRP78 positive areas after staining contiguous sections with an antibody against GRP78, and ER chaperone.
|
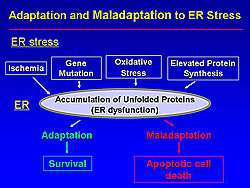
The classic functions of ER include the folding of secretory and membrane proteins, regulation of storage and release of calcium ions, and the synthesis of phospholipids and steroids. In addition, now the ER is also considered as a signal transducer organelle that determines cell survival or cell death.
A recent study demonstrated that various stimulants such as ischemia, gene mutation, oxidative stress, and elevated protein synthesis cause ER dysfunction (Figure 1). Adaptive survival signals are initiated when the ER is compromised by these stressors. Notably, the ER initiates apoptotic cell death when the ER stress is excessive or prolonged.
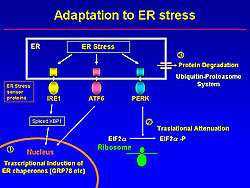
Adaptation to ER stress
Three transmembrane proteins sense the ER stress (IRE1, ATF6, PERK), and these activate three adaptive pathways (Figure 2). One, the transcriptional induction of ER chaperones to help protein folding, and thus increasing expression of GRP78, a marker of ER stress. Two, the translational attenuation of protein synthesis, which blocks new protein synthesis. Three, the degradation of accumulated protein via the ubiquitin-proteosome system. These three mechanisms work cooperatively to reduce ER stress.
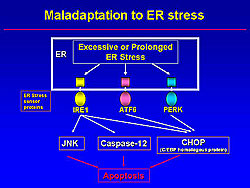
However, when the ER stress is extensive or prolonged, apoptotic signals, including JNK, Caspase-12, and CHOP (C/EBP homologous protein) are activated (Figure 3). Among them, CHOP is a specific molecule of ER-initiated apoptosis.
|
|
Working hypothesis of ER-initiated apoptosis in heart failure
|
|||
|
|||
|
|||
|


Thus, Minamino and colleagues hypothesized that oxidative stress and increased protein synthesis in hypertensive hearts causes ER stress, and when this ER stress is excessive or prolonged, ER-initiated apoptosis occurs and contributes to the development of heart failure (Figure 4).
In the present study, this group tested their hypothesis using human heart samples, mouse hypertrophic and failing hearts, and cultured rat neonatal cardiac myocytes.
In the patients with heart failure, GRP78 and CHOP were markedly induced compared to the control hearts. This finding suggests that ER stress is induced and ER-initiated apoptotic signals are activated in failing human hearts.
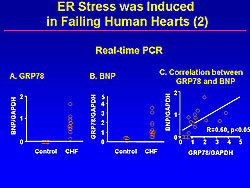
The expression of GRP78 and BNP was increased in failing human hearts, using real-time PCR. Interestingly, a positive correlation between GRP78 and BNP was found, suggesting that ER stress may induce cardiac dysfunction (Figure 5).
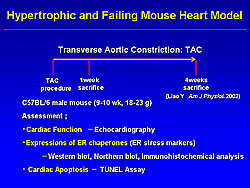
In the hypertrophic and failing mouse model, created by transverse aortic constriction (TAC), cardiac enlargement was induced 1 week after TAC. At 4 weeks, more cardiac enlargement and severe lung congestion were induced. Echocardiographic findings revealed cardiac hypertrophy at 1 week and reduced contractility at 4 weeks after TAC, corresponding to the gross anatomical findings (Figure 6). On Northern Blot analysis, mRNA levels of ER chaperones were increased at 1 and 4 weeks after TAC. Also, GRP78 was induced in these hypertrophic and failing hearts. These findings indicate that pressure overload by TAC induced prolonged ER stress.
The relationship between cardiac apoptosis and apoptotic signaling from ER was assessed, showing that the number of TUNEL-positive cells was increased in the heart at 4 weeks after TAC, from nearly 0 in the sham rats to about 0.4% (p<0.05). CHOP was markedly induced at 4 weeks—timing that is very consistent with cardiac apoptosis (Figure 7). Caspase12 or P-JNK was not activated. These findings suggest the possibility that prolonged ER may induce cardiac apoptosis via a CHOP-dependent pathway.
A pharmacologic ER stress inducer (tunicamycin, thapsigarging) induced the ER chaperones. Furthermore, these agents increased the TUNEL-positive cells in cardiac myocytes.
Pharmacologic ER stress increased CHOP protein levels in cardiac myocytes. Importantly, pharmacological-induced apoptosis was partially but significantly attenuated by the knock-down of CHOP protein. These findings suggest that ER stress induced cardiac apoptosis partially by the CHOP-dependent pathway.
Summary
|
ER-stress and ER-initiated apoptosis signals were activated in failing human hearts (Figure 8). Interestingly, there was a positive correlation between BNP and GRP78. Pressure overload of the mouse heart induced prolonged ER stress. Apoptosis appeared along with CHOP expression in failing hearts. ER stress induced apoptosis partially via a CHOP-dependent pathway in cardiac myocytes.
Nifedipine Prevents the Progression of Coronary Atherosclerosis More Effectively than ACE-inhibitors in Hypertensive Patients with CADYoshiki YuiKyoto University
|
The Japan Multicenter Investigation for Cardiovascular diseases (JMIC-B) study investigated the effect of nifedipine retard (20-40 mg/day) and ACE inhibitors on the long-term prognosis in patients with hypertension and coronary artery disease (CAD) using the PROBE design and a 3-year follow-up.
No significant difference in the primary endpoint of the cumulative event rate between the groups was found in JMIC-B. No significant difference was found in the relative risk for cardiovascular events between the groups.
The QCA study of JMIC-B investigated the effect of nifedipine retard and ACE inhibitor on the progression and regression of coronary atherosclerosis. A total of 210 patients (108 nifedipine, 102 ACE inhibitor) who underwent coronary angiography (CAG) were enrolled in this QCA study. After 3 years, follow-up CAG was performed. A total of 162 patients (83 nifedipine, 79 ACE inhibitor) were eligible for QCA criteria.
The CAG was performed after intracoronary administration of ISDN, and the angiograms were analyzed in the core laboratory using the Cardiovascular Measurement System. The coronary tree was divided into 13 segments, according to the AHA classification. Narrowing within the segments was analyzed, and calculations for the average minimum lumen diameter performed.
The changes found on QCA were defined as:
- Progressor, a patient with ≥1 lesion worsening by ≥0.4 mm, or a complete occlusion, or a new lesion
- Stable, a patient with no lesions worsening or improving by at least 0.4 mm
- Regressor, a patient with ≥ 1 lesion improving by ≥0.4 mm, or no lesions worsening by ≥ 0.4 mm
- New lesion, ≥21% diameter stenosis reducing the lumen diameter by ≥0.4 mm.
The primary endpoint of the QCA study was the change in minimum lumen diameter (MLD) and % diameter stenosis (DS) in each patient. The secondary endpoints were the number of each type of patient (progressor, regressor, stable), and the number of new lesions and patients with new lesions.
At baseline, the groups were similar. In the nifedipine and ACE inhibitor groups, respectively, the baseline blood pressure was 145/81 mmHg and 144/80 mmHg; final blood pressure 135/74 mmHg and 133/76 mm Hg; and the total cholesterol in the observation period and after 36 months was 5.1 mmol/L.
For the primary endpoint, a decrease in MLD was observed for all segments in the ACE inhibitor group, but an increase in MLD was seen with nifedipine (-0.12 mm vs +0.02 with mm, respectively; p=0.002). This same trend was seen for the segments of the coronary lesion (-0.01 mm vs +0.12 mm, respectively; p=0.016).
In the nifedipine group, compared to the ACE inhibitor group, fewer patients were progressors (31 vs 44 patients; p=0.019) and fewer patients had new lesions (22 vs 33 patients, p=0.040).
Conclusions
The CAMELOT study using IVUS to evaluate the suppression of the progression of coronary atherosclerosis compared to baseline showed progression in the placebo group, a trend toward progression in the enalapril group, and no progression in the amlodipine group.
Quantitative coronary angiogram in the JMIC-B study showed that both nifedipine and ACE inhibitors may be effective to suppress the progression of coronary atherosclerosis. Nifedipine may be an effective treatment for mild to moderate coronary stenosis. However, QCA data must be complemented by IVUS. The evaluation of the character of the vessel wall by IVUS is necessary for the understanding of vessel arteriosclerosis.
|
|
| Copyright © 2005 Japanese Circulation Society All Rights Reserved. webmaster@j-circ.or.jp |