 |
|
|
|
IS154
Keynote Lecture
Cell Cycle Regulation and Myocardial Regeneration? |
|
Harold S. Bernstein, M.D.,
Ph.D.
Cardiovascular Research
Institute
University of California, San Francisco, USA
San Francisco, CA, USA |
|
|
|
|
|
|
|
|
Liansen Liu, PhD reviewed work in the laboratory of Harold
Bernstein that has led to the first evidence that the
human protein hCdc5 is a site-specific DNA binding protein
capable of forming high affinity complexes with specific
genomic sequences. Further work is required to define
the mechanisms by which hCdc5 regulates mitotic entry
and its possible use to enhance myocyte proliferation.
Cumulative death of cardiac myocytes is the cellular basis
underlying acute myocardial infarction and myocardial
insufficiency, and prognosis is directly correlated with
the amount of viable cardiac tissue mass.
|
PAGE
TOP
Cardiac myocyte
cell cycle |
|
Cardiac myocytes begin to withdraw from the cell
cycle in the second half of gestation, continue
to decline through late gestation, and cease proliferating
within weeks of birth. This phenomenon of cell cycle
withdrawal has been studied extensively in rodents,
where data shows that in the first two weeks after
birth the percent of myocytes with detectable DNA
synthesis drops to zero. Thereafter, cardiac myocytes
demonstrate no innate capacity for regeneration.
Can a myocyte can be engineered that will both
function and proliferate? Work in Bernstein's laboratory
is testing their hypothesis that myocytes can be
manipulated to accomplish this. Existing descriptions
of myocyte-derived cell lines that proliferate while
maintaining a cardiac phenotype suggest that cell
division and myocyte functions are not necessarily
exclusive. The feasibility of creating functioning,
multiplying myocytes is based on several observations:
- cardiac myocytes in the fetus are capable of
some differentiative function while maintaining
the ability to proliferate
- a cell line, derived the from the atrial myocytes,
has recently been described that continues to
proliferate in culture while maintaining many
of the features of cardiac myocytes
- micrographic figures of myocytic mitosis in
patients with end-stage heart failure suggest
that some mechanism is present that allows a few
cells to return to division.
|
|
PAGE
TOP
Transcriptional
reprogramming for myocytes |
|
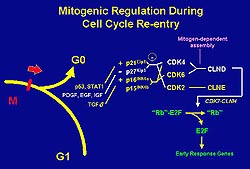 |
Figure
1. Schematic of mitogenic regulation. The cyclin
dependent kinase (CDK) complex is the primary
driver of this cell cycle. Transcription is activated
by CDK4 and CDK6, which turn on the early response
genes essential for cell-cycle progression. In
the myocyte, the transcription activator MyoD
upregulates the CDK inhibitor p21, which causes
the inhibition of CDK4 and cyclin D complex (CLND),
thus causing the cell cycle withdrawal necessary
for myocyte differentiation. (Harold Bernstein
April 2000)
Click
to enlarge |
 |
Figure
2. CDC2-cyclin B (CLNB) comprises the cyclin dependent
kinase complex that drives the G2/M transition
of the cell cycle. This complex is regulated through
phosphorylation and de-phosphorylation events
carried out by Wee1 and Myt1 kinases, Cdc25C phosphatase,
and the CDK7-cyclin H (CLNH) complex. Although
upstream regulators of these phosphatases and
kinases are known, the transcriptional regulation
of these events is not well understood. (Harold
Bernstein April 2000)
Click
to enlarge |
|
Although most myocytes remain quiescent from birth
on, a mechanism must exist to activate their proliferation.
The work in Bernstein's laboratory addresses the
series of events that comprise the biology of myocytes,
especially as it relates to their transcription
and regulation. The group is exploring methods to
manipulate this division cycle.
Ordinary cells duplicate in the S phase and
separate in mitosis. This cell division requires specific, appropriate conditions for genetic
material to be duplicated completely and without
error, and for the mitotic spindle to be appropriately
assembled. The primary oscillator driving the cell
cycle is the cyclin dependent kinase (CDK) complex
(Fig. 1). Cell cycle transcription and regulation
is dependent on various cell stimuli and is regulated
by specific CDK inhibitors. Transcription is activated
by CDK4 and CDK6, which turn on the early response
genes essential for cell-cycle progression. In the
myocyte, the transcription activator MyoD upregulates
the CDK inhibitor p21, which causes the inhibition
of CDK4 and cyclin D complex (CLND), thus causing
the cell cycle withdrawal necessary for myocyte
differentiation.
Animals that overexpress the cyclin D complex have
an increased number of myocytes. Recent research
has sought to bypass the CDK control. Cultured myocytes
overexpressing E2F reentered the cell cycle but
then died. Transcriptional reprogramming with E2F
bypasses the mitogenic requirement for cell cycle
re-entry, preventing the apoptotic process. E1B
protects against apoptosis by an unknown mechanism,
but cells are then arrested in G2.
Bernstein's group has proposed that similar transcriptional
reprogramming might bypass requirements for G2/M
transit in myocytes. Cardiac myocytes re-enter the
cell cycle and synthesize DNA with overexpression
of G1/S-specific transcription factors. These cells
remain blocked in G2, however, and failed to enter
mitosis. These cells get stuck in G2, but G2/M transit
requires the coordinated expression of many genes, and
little is known about their transcriptional regulation.
Figure 2 summarizes the current thinking about G2/M
regulation. Transcriptional reprogramming may bypass
requirements for G2/M.
|
|
PAGE
TOP
First
transcriptional
regulator defined |
|
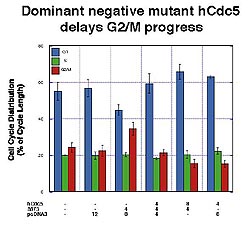 |
Figure
3. G2/M transit is substantially delayed in the
presence of the dominant negative mutant hCdc5
containing only the amino terminal DNA binding
domain. Overexpression of wild-type hCdc5 competed
against this effect. (J Biological Chemistry 1998;273(8):4666-4671)
Click
to enlarge |
|
Identification of G2/M regulators would significantly
advance the understanding of this portion of the
cell cycle, and provide new reagents for manipulating
myocytes. Several years ago, Bernstein's group described
their cloning and analysis of the human protein
Cdc5 (hCdc5) in mammalian cells. This protein was
remarkable because of its:
- homology to cell cycle regulators in yeast
- evolutionary conservation
- putative DNA binding domain
- existence in the cytoplasm of quiescent cells,
followed by its nuclear translocation and phosphorylation
with cell cycle re-entry.
Their work has also shown that overexpression of
hCdc5 alters cell cycle distribution in asynchronous
cultures; overexpressing hCdc5 can accelerate G2/M
transit and S phase. Cells in which hCdc5 was inducibly
overexpressed spend approximately the same amount
of time as controls in the G1 phase, but less in
S phase and significantly less time in G2/M transit.
To corroborate this finding, they created dominant
negative mutant hCDc5 that contained only the amino
terminal DNA binding domain. G2/M was substantially
delayed in the presence of the mutant, but overexpressing
wild-type hCdc5 competed against this effect (Fig.
3). This provided further evidence that hCdc5-like
proteins are positive regulators of mitotic entry.
Thus, overexpression of hCdc5 shortened G2, while
a dominant negative mutant lacking its activation
domain delayed mitotic entry_implicating hCdc5 as
the first defined transcriptional regulator of G2/M
in mammalian cells.
|
|
PAGE
TOP
DNA binding
sequence for hCdc5 identified |
|
Elucidating the mechanism by which hCdc5 regulates
cell division has been the focus of studies by Bernstein's
group. In response to proliferative growth factors,
hCDc5 undergoes nuclear transformation and phosphorylation,
including additional phosphorylation in the nucleus
that regulates hCDc5's interaction with downstream
genes. hCdc5 appears to contain a domain capable
of activation transcription, which Bernstein's group
has sought to identify.
A preferential DNA binding site for hCdc5 was identified
in their experiment looking for support for hCdc5's
role as a binding protein. By performing cyclic
amplification and selection for targets for a pool
of random oligonucleotides, they identified a 12
base-pair sequence that binds to hCdc5 with affinity
and specificity. The results showed that the interaction
between hCdc5 and the DNA is specific, and that
hCdc5 and the DNA had an affinity to the degree
of other helix DNA binding proteins.
To determine if the specific binding site is present
in the human genome, a yeast-based screening strategy
was devised. They identified 12 clones that appeared
to have the sequence that can bind to hCdc5, using
a library of genomic DNA fragments and incorporating
a series of counter screens. Several of these clones
contained a sequence of base pairs very similar
to the sequence they had previously identified.
Presumably this sequence represents the regulatory
elements of downstream genes for hCdc5. They also
showed that the uncharacterized C-terminal of hCdc5
might contain negative regulating elements.
Thus, a high-affinity consensus sequence that
binds hCdc5 through its HTH domain has been identified
by Bernstein's group. They also showed this interaction
occurs in vivo. Although they have searched the
human genome database for a corresponding sequence
in genes already characterized, this has proven
difficult because this sequence presents in very
small numbers in the human genome. In summary:
- Cdc5 is a positive regulator of G2/M in mammalian
cells
- Cdc5 is phosphorylated and translocates to the
nucleus in serum-stimulated cells
- Cdc5 binds specifically and with high affinity
to a consensus, double-stranded DNA sequence
- Consensus binding sites for Cdc5 are present
in low numbers in the human genome.
|
|
PAGE
TOP
Possible
use to enhance myocyte proliferation |
|
Defining mechanisms through which hCdc5 regulates
mitotic entry may facilitate its use in reagents
for manipulating the cell cycle in nondividing cardiac
myocytes. Cdc5 is likely regulated through mitogen-activated
signaling pathways. Phosphorylation mutants may
provide reagents to manipulate the mammalian cell
cycle. Cdc5 can act as a site-specific DNA binding
protein. Cdc5's function in G2/M is likely mediated
through DNA-protein interactions. Cdc5 targets are
present in the human genome. Their identification
will delineate the pathways by which Cdc5 regulates
mitotic entry.
|
|
PAGE
TOP
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2000
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|