 |
|
|
|
IS155
Genetic
Dissection of Cardiac Life and Death Cascades |
|
Michael D. Schneider, M.D.
Baylor College of
Medicine
Houston, TX, USA |
|
|
|
|
|
|
|
|
The concept of heart failure as a myocyte-deficient
disease has been the focus of research in Schneider's
laboratory. This concept is supported by the fact that
the "post-mitotic phenotype" (loss of cell proliferative
capacity after birth) and the chronic loss of cardiac
muscle cells in heart failure via apoptosis limit the
potential to restore cardiac pump function through an
increase in the number of cardiac muscle cells.
Two approaches have been taken in their
laboratory. One, to override or bypass mechanisms for
"irreversible" cell cycle exit. Viral gene delivery of
exogenous activators, notably the adenoviral protein E1A
and E2F-1, and gene deletion to remove endogenous cell-cycle
inhibitors, focusing on the retinoblastoma gene product
Rb, and the cyclin dependent kinase (CDK) inhibitors p21
and p27 have been studied. Two, to reduce cell death from
apoptosis directly. Efforts to identify proteins that
trigger this cascade and efforts to inhibit the triggers
of apoptosis itself, involving work in transgenic mice
overexpressing the anti-apoptotic protein Bcl-2 in myocardium,
have been undertaken. TAK1, a TGF-beta activated kinase,
an apoptotic trigger has been studied.
Schneider reviewed work in their lab showing
that irreversible cell cycle exit can be overridden by
viral delivery of the exogenous activators E1A or E2F1.
By deleting the endogenous inhibitors Rb, p21, and p27
at least partial override can be achieved. However, both
of these interventional approaches result in the accumulation
of cells at the G2/M boundary, an important gap in present
knowledge.
In their efforts to reduce myocyte death
in apoptosis, they have shown that a TGF-beta activated
kinase, TAK-1, is activated in vivo as a delayed response
to mechanical load. This kinase is sufficient to produce
the molecular, morphological, and functional hallmarks
of cardiac hypertrophy in vivo, and is a logical target
for pharmacological or genetic countermeasures. By using
an inhibitor of apoptosis itself, Bcl-2, they have been
able to reduce infarct size by 50% in mice, at high levels
protein expression. No effect has been seen at lower levels
of expression, coupled with an 80% loss of the protein
through degradative mechanisms, suggesting that engineered
forms of Bcl-2 might be more appropriate to try to reduce
cell death from apoptosis therapeutically.
|
PAGE
TOP
|
|
|
The retinoblastoma gene product (Rb) is developmentally
regulated in the myocardium, as shown by Western
blot analysis. Little or none is expressed in midgestation
in myocardium and Rb is the predominant pocket protein
expressed in the adult. p107 has a reciprocal pattern
of expression. p130 is expressed at all three ages.
This is noteworthy because it had been postulated
that functional differences between Rb and p107
and the developmental regulation of these proteins
was responsible for cell cycle exit and the irreversible
loss of cell cycling in post-mitotic skeletal muscle.
That model is somewhat paradoxical, since Rb is
a protein whose function is totally reversible upon
protein phosphorylation. Work in their laboratory
has sought a model in which irreversible cell cycle
exit might be constructed by the developmental regulation
of Rb, versus p107, in concert with other inhibitors,
such as p21 and p27, abundant in the post-mitotic
heart but expressed at low or negligible levels
in cardiac proliferation.
|
 |
Figure
1. The signaling cascade for mitogen signal transduction.
See text for details. (Heart Development, Ed Harvey
RP and N Rosenthal. Academic Press, Tokyo.)
Click
to enlarge |
|
The signaling cascade for mitogen
signal transduction has been elucidated through
their work with the adenoviral E1A (Fig.1). Mitogens
activate D-type cyclins whose targets are CDK4 and
CDK6 whose essential substrates are pocket proteins.
Pocket protein phosphorylation then disinhibits
E2F-dependent gene transcription. This results in
activation of cyclins E and A and other E2F-induced
genes, and the increase in CDK2 activity necessary
for entry into S phase and DNA synthesis.
In contrast, forced cell cycle re-entry
in cardiac myocytes, triggered by E2F-1 to bypass
pocket proteins, remain sensitive to blocks to CDK2
activity, including p21 or dominant negative CDK2.
This inactivates pocket proteins using E1A, resulting
in DNA synthesis that, surprisingly, proved to be
independent of an increase in CDK2 activity. S phase
entry driven by E1A was resistant to p21 and dominant
negative CDK2 inhibitors, and occurred at levels
of CDK2 activity no greater than those seen in serum-starved,
growth-arrested post-mitotic cells. Four potential
mechanisms for these observations are being studied:
1) E1A itself results in substrate activation or
inactivation of a substrate through protein-protein
interactions, 2) alternative E2F family members,
3) other pocket protein targets, and 4) other E1A
targets, including p400, a poorly characterized
protein known to bind to regions of E1A necessary
for CDK2 independent effects.
However, this work does not show whether
the endogenous genes play a role in cell cycle exit.
The loss of p21 resulted in a significant delay
in cell cycle exit in cardiac myocytes, as shown
by flow cytometry in newborn mice (wild type, missing
p21 or p27, or both). A 2-fold increase in the number
of cells with S or G2M DNA was found. However, this is a delay in cell cycle exit,
not a permanent ability to continue cycling, as
animals even 14 days of age have negligible DNA
synthesis in myocardium. In p27 null mice, p27 has
a roughly 2-fold larger effect than loss of p21.
Mice lacking even one copy of p27 are sufficient
for effects as large as those seen in p21 null mice.
Interestingly, with the combined deletion of p21
and p27, DNA synthesis is shut down and little or
none is seen even at 14 days of age. Roughly 30%
of these cardiac myocytes have DNA content greater
than G0/G1 at this stage in the newborn.
|
|
PAGE
TOP
|
The nominal gold standard gene deletion
approach for determining protein function in vivo
could not used with Rb. The conventional Rb knockout
results in embryonic lethality in midgestation with
mammoth defects in hematopoiesis and neurogenesis.
Thus, technologies for conditional gene deletion
using the Cre/lox system have been used: when a
gene is tagged in innocuous regions, such as within
its introns, with 34-base-pair motifs known as loxP
sites and the Cre recombinations are introduced,
DNA recombination is triggered between the loxP
sites.
Schneider's lab has used a Cre-dependent
reporter gene as a knock-in to the ubiquitously
transcribed rosa26 locus. In the absence of Cre,
little or no recombination as measured by activation
of a Cre-dependent lacZ gene is seen. When these
mice are mated to mice containing Cre-recombinase
driven by the alpha-myosin heavy chain promoter,
nearly uniform recombination is seen in atrial and
ventricular myocytes by 10.5-11 days gestation.
The Cre-dependent promoter is sometimes dismissed
in the literature as being expressed predominantly
in the embryonic atria and only comes up in the
ventricle after birth. But, the levels of expression
seen in the future ventricle at the linear heart
tube stage are sufficient to trigger recombination
in nearly all ventricular myocytes.
A recombination-specific gene product
appears in the myocardium of the crossed alpha-myosin
heavy-chain Cre mice and lox-p Rb mice. There is
loss of the intact Rb allele, almost complete loss
of the Rb protein from the myocardium, and spontaneous
DNA synthesis even under basal conditions, as shown
by immunoperoxidase staining and two-color immunofluourescence.
The absolute frequency for this event is low, roughly
one cell in a thousand, similar to that reported
in mice myocardium overexpressing cyclin D1. Thus,
other inhibitors are likely present either in series
with Rb, such as p21 and p27, or in parallel in
with Rb, notably p130, the other pocket protein
that coexists with Rb in myocardium.
This work is the first demonstration
of an essential function for pocket proteins in
cardiac growth control in vivo, they believe. This
is based on the observation that when the relatively
innocuous cardiac-restricted Rb knockout is made
into the conventional, totally innocuous p130 knockout,
about a 2-fold increase in heart size occurs between
4 and 8 weeks of age. Promiscuous reactivation of
a number of cell-cycle regulators, including cyclin
B, usually transcriptionally suppressed in adult
heart results. The CDK inhibitor knockout and Cre
knockout work is ongoing.
|
|
PAGE
TOP
Cardiac
development model |
|
A cardiac-specific knockout of the
bone morphogenic protein (BMP) was developed. BMP
is a member of the transforming growth factor beta
(TGF-beta) superfamily that has been implicated,
through circumstantial evidence and explant model
systems, as potential regulators of cardiac development.
Komuro reported recently that BMP signaling was
important for cardiac myogenesis in p19 embryonic
carcinoma cells in the mouse model. The role of
BMPs or their type-1 receptor ALK3 can not be studied
in myocardium by conventional deletion because of
lethality at gastrulation or other early stages
before heart formation in BMP2, BMP4, and ALK3.
In their lab, no survivors to late
gestation were seen when mating alpha MHC-Cre mice
to mice loxP tagged for the ALK3 gene. Marked resorption
even by embryonic day 15 and internal hemorrhages
by embryonic day 13 were seen. All mice had large
atrial septal or ventricular defects. In some animals,
the endocardial cushion was present but hypocellular
and disorganized. In others, the endocardial cushion
was absent and a single four-chambered heart observed.
At least a subset of the animals was defective for
expression of NKX2.5, the cardio-restricted homeobox
gene; mechanistic studies are ongoing.
A marked increase in apoptosis in
the septum was caused by the absence of BMP signaling
through ALK3. Thus, they think a BMP signal, directly
or indirectly, is responsible for cell survival
in cardiac morphogenesis. A number of genome-wide
technologies to identify the BMP-dependant genes
both for survival in septum and for normal morphogenesis
involving the atrioventricular cushion is being
pursued by their lab.
|
|
PAGE
TOP
|
| |
BMP signaling is known to occur through
at least two pathways. One involves Smad transcription
factors. A different set of Smad transcription factors
is implicated in signaling for TGF-beta itself.
Both the BMP and TGF beta pathways have been reported
to involve signaling through MAP kinases,
including a TGF beta-activated kinase (TAK), which
lies upstream of JNK and p38.
|
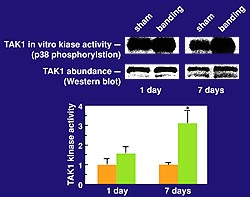 |
Figure
2. TAK1 activity in myocardium in vivo is upregulated
as a delayed response to aortic banding. Activation
of a kinase as a delayed response following pressure
overload is seen, consistent with its involvement
in time-dependant autocrine-paracrine mechanisms
Upregulation of TAK levels is not seen. (Nature
Medicine 2000;6(5):556-562.)
Click
to enlarge |
|
TAK1 activity in myocardium in vivo
is upregulated as a delayed response to aortic banding,
as shown in their lab by immune complex kinase assays
in which endogenous TAK1 was immuno-precipitated
and incubated with its substrates (Fig.2, upper
panel). Upregulation of TAK levels is not seen.
Activation of a kinase as a delayed response following
pressure overload is seen, consistent with its involvement
in time-dependant autocrine-paracrine mechanisms
(Fig 2, bottom panel).
In tissue culture, the TAK1 signal
was sufficient to mimic the effect of TGF beta on
the skeletal alpha actin promoter, a marker of hypertrophy.
Importantly, TAK1 kinase activity was necessary
for TGF-beta signaling and was completely extinguished
by dominant negative TAK1. This process involved
the p38 pathway, not JNK, and the protein ATF6,
recently implicated in adrenergic and endothelin-dependant
signaling to other serum response factor-dependant
cardiac promoters.
Schneider's lab created transgenic
mice overexpressing TAK1 in the myocardium that
have the same 3- to 4-fold increase in TAK-1 activity
seen with pressure overload. A 40-50% increase in
heart size by ten days of age, associated with fulminant
mortality, resulted. No F1 animal survived beyond
15 days of age. The F0 founders survived for three
to six months, ultimately dying of hypertrophy and
heart failure. Mosaicism in the founder mice accounts
for the difference between F0s and F1s. The TAK1
transgenic mice had 1) myocyte enlargement, 2) myocyte
drop out and fibrosis, 3) reactivation of fetal
gene expression, 4) a decrease in systolic and diastolic
function, and 5) a marked increase in apoptosis,
as shown by TUNEL staining. No apoptosis in wild-type
animals and the expected 2-5% prevalence of apoptosis
in mammary epithelium as a positive control was
seen.
Thus, they believe TAK1 is activated
in vivo as a response to mechanical load. It signals
via p38 to ATF6 and other downstream proteins, and
is sufficient to trigger the molecular, morphological,
and functional hallmarks of cardiac hypertrophy.
The early mortality makes it especially attractive
as a potential test bed for counter measures aimed
at cardiac apoptosis or other components of the
heart failure phenotype.
|
|
PAGE
TOP
|
A cardiac-specific Bcl-2 transgenic
mouse model has been created in their lab. Bcl-2
is the prototype of a family of pro- and anti-apoptotic
proteins that act by preventing the mitochondrial
transitions that couple upstream apoptotic signals
to downstream effector pathways, including downstream
caspase activation as part of the apoptotic cascade.
Human Bcl-2 was shown to be expressed in the 4-copy
and 14-copy lines. The endogenous protein is not
altered in expression in the presence of the transgene,
and the levels of are increased about 3-fold in
the 4-copy line and 7-fold in the 14-copy line.
A marked reduction in infarct size
associated with rescue of systolic and diastolic
function was shown after one hour of ischemia and
24 hours of reperfusion in the high-copy Bcl-2 line.
Rescue was not seen in the lower copy line, suggesting
a dose-dependant relationship. The Bcl-2 protein
is markedly downregulated early with ischemia-reperfusion
injury, and may potentially explain the all-or-nothing
effect seen. In the early hours of ischemia-reperfusion
there is an 80% loss of protein, which is partially
recovered within 24 hours.
Two reported mechanisms for Bcl-2
degradation are likely active in ischemia-reperfusion.
Bcl-2 is downregulated through inhibition of MAP-kinase-dependent
phosphorylation, which can be triggered by TNF-alpha,
among other agonists. Bcl-2 is also a reported target
for degradation via proteolysis by caspases. Their
second-generation Bcl-2 mice contain a phosphomimetic
mutation at the sites for MAP-kinase phosphorylation;
a reportedly constitutively stable mutation in tissue
culture, even under TNF-alpha stimulation. Third-generation
mice with caspase cleavage site mutations are underway.
They expect that mutations of Bcl-2,
engineered forms of Bcl-2 resistant to the two degradation
pathways, may be capable of providing cardioprotection
at lower basal levels of expression. And, may be
resistant to the degradation seen with ischemia-reperfusion
in injury, and ultimately may be the forms of Bcl-2
selected for translational studies using viral gene
delivery to the myocardium.
|
|
PAGE
TOP
Report
Index | Previous Report
| Next Report
Scientific
Sessions | Activities
| Publications
Index
Copyright © 2000
Japanese Circulation Society
All Rights Reserved.
webmaster@j-circ.or.jp
|
|