|
|
||||||
|
|||||||
|
|||||||
|
|||
|
|||
Role of T Cells in Autoimmune MyocarditisMakoto KodamaNiigata University Graduate School of Medical and Dental Sciences
|
Collateral damage from cardiac viral infections and autoimmune myocardial injury are principle pathologic mechanisms underlying inflammatory myocardial disease and non-ischemic and non-genetic cardiomyopathy. Dr. Kodama, Niigata University Graduate School of Medicine and Dental Sciences, investigated inflammatory mechanisms of autoimmune myocarditis using a cardiac myosin-induced experimental autoimmune myocarditis (EAM) model.
EAM is an animal model of human giant cell myocarditis. Rat EAM is characterized by 100% morbidity and significant mortality from heart failure. The goals of this investigation were to: 1. Learn how cardiac myocytes are destroyed by cardiac myosin-specific T cells, and 2. Determine why some EAM animals recover spontaneously even though large amounts of antigen are present in the heart.
T cells in EAM rats are activated by a costimulatory signal at the time of antigen recognition that includes the CD28 molecule. EAM rats were injected with the cytotoxic T cell associated antigen-4 (CTL-4) gene. CTL-4 inhibits the action of CD28. On day 17, histologic evaluation showed that induction of EAM was suppressed.
|
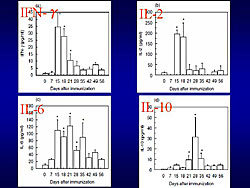
Th1/Th2 cell balance in EAM was analyzed by flow cytometry. The proportion of Th1 cells, which produce IFN-γ, increased with the onset of EAM, on day 14. During the active phase, Th2 cells, which produce IL-4, gradually increased, overcoming Th1 cells on day 28. During this phase, there was resistance to active immunization. By the chronic phase, the Th1/Th2 cell balance returned to the basal level. Serum levels of Th1 and Th2 cell-induced cytokines were measured (Figure 1). IFN-γ and IL-2 increased in the acute phase and decreased in the early recovery phase. IL-10 increased at the early recovery phase.
Other studies showed that blocking TNF, IL-4, IL-12, and IL-1 receptor (IL-1R) suppress induction of EAM. Inhibition of IL-2 and IFN-γ exacerbates EAM. Therefore, cardiac myosin-induced murine EAM is a Th2 dependent disease. In vivo gene therapy with IL-10 DNA before and after antigen sensitization showed that IL-10 attenuates EAM, but the effect is incomplete. IL-10 decreased histamine, reduced the number of mast cells, and suppressed the growth and degranulation of mast cells, implying that mast cells contribute to development of EAM.
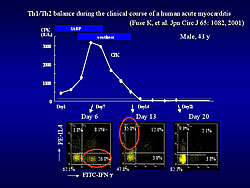
In a human study, the kinetics of Th1/Th2 cell balance in peripheral blood from a patient with acute myocarditis was similar to that in rat EAM (Figure 2). Analysis of Th1/Th2 cell balance is valuable in diagnosis and understanding of human myocarditis.
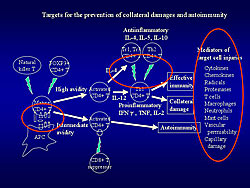
These studies demonstrate the existence of several therapeutic targets to prevent collateral damage and autoimmune myocardial injury (Figure 3).
|
|
Viral Infection and AutoimmunityAkira MatsumoriKyoto University Graduate School of Medicine
|
Dr. Akira Matsumori, Kyoto University Graduate School of Medicine, reviewed studies investigating the relationship of anti-heart autoantibodies with the pathogenesis of myocarditis and cardiomyopathy. In the past 20 years, various autoantigens of cardiac autoantibodies have been found in patients with dilated cardiomyopathy (DCM), including sarcolemmal proteins, mitochondrial proteins, heat-shock proteins, and membrane receptors.
|
||
|
Coxsackievirus B infection induces myocarditis associated with anti-myosin antibody. In previous studies (1993), Matsumori showed that viral (EMCV)-induced murine myocarditis resulted in cardiomyopathy and heart failure. Autoantibodies from mice with EMCV myocarditis increased the intracellular free Ca2+ concentration through the activation of Ca2+ permeable cation channels, possibly through the action of a soluble intracellular messenger.
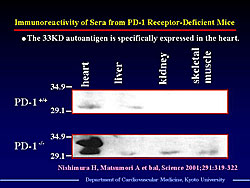
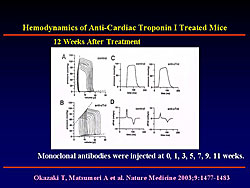
In 1999, Matsumori and colleagues reported that mice deficient in programmed cell death-1 (PD-1) immuno-inhibitory coreceptor developed autoimmune cardiomyopathy and heart failure. PD-1 is expressed on activated T and B cells and negatively regulates antigen-receptor signaling. Subsequent studies demonstrated that PD-1 deficient mice develop high-titer autoantibodies against a heart-specific 33 KD protein (Figure 1). This protein was extracted and identified as cardiac troponin I (cTnI). Monoclonal antibody to cTnI was injected into wild-type mice. At 12 weeks after injection, the mice showed suppressed cardiac function with increased systolic and diastolic volume, reduced ejection fraction, and decreased dP/dt (Figure 2). This finding demonstrated that antibody to cTnI directly induced myocardial injury. Western blot analysis of sera from patients with DCM has demonstrated the presence of anti-cTnI antibody.
Matsumori reviewed results of several studies of cardiac autoantibodies in patients with viral infection. A study of 112 patients with active lymphocytic myocarditis treated with immunosuppressive therapy reported presence of cardiac autoantibodies in 90% of responders and in none of the nonresponders. The beneficial effect of immunosuppression suggests immune-mediated damage in these patients.
In HCV patients with antimitochondrial antibodies, two-thirds had associated Sjogren’s syndrome or systemic sclerosis and a high frequency of multiple autoantibodies. In a study of the impact of HIV co-infection on the presence of autoantibodies in patients with HCV, 71% of co-infected patients had anti-smooth antibody (ASMA). Patients with ASMA showed a trend toward more bridging fibrosis or cirrhosis. Laboratory studies of autoimmune markers in patients with HCV infection discovered the presence of several autoimmune antibodies. Rheumatoid factor was found in 45-70%, cryoglobulins in 40-55%, antinuclear antibodies in 10-20%, Anti-smooth muscle antibodies in 7-27%, anti-thyroid antibodies in 5-10%, and antimitochondrial antibodies in 1%.
Matsumori concluded that autoimmunity may play an important role in the pathogenesis of viral heart diseases. Clarification of the role of autoimmunity may contribute to the development of new therapies for viral myocarditis.
Immune Modulation and Chronic Heart FailureStephen AnkerCharite Campus Virchow-Klinikum
|
Progression of chronic heart failure (CHF) is associated with immune dysregulation and increased inflammatory cytokines. Consequences of inflammatory cytokines in CHF include cardiac dysfunction, insulin resistance, endothelial dysfunction, catabolic metabolism, apoptosis with tissue wasting and cachexia, and poor survival. Dr. Stephen Anker of Applied Cachexia Research and Charite Campus Virchow-Klinikum, Germany, reviewed therapeutic approaches to anti-inflammatory therapy in CHF.
Clinical trials of anti-inflammatory therapy for CHF focused on targeting tumor necrosis factor (TNF) have failed to show benefits. Therapy needs to be targeted to CHF patients with inflammation or, perhaps, those with wasting syndrome. Additionally the focus should be on shifting the Th1-Th2 cell balance rather than eliminating a single cytokine such as TNF.
Neurohormones and bacterial endotoxins may play a role in CHF. In the context of neuroendocrine activation, responsiveness to small amounts of endotoxin may increase. Plasma lipoproteins protect against endotoxins by forming micelles around them, preventing interaction with mononuclear cells. A study showed that whole blood of CHF patients with higher serum lipids was less responsive to administration of lipopolysaccharide (LPS). Epidemiological studies reported that patients with ischemic CHF with higher total cholesterol and LDL levels live longer, and those with low cholesterol have higher inflammatory cytokine levels.
Anker and colleagues studied the effect of nutrition therapy in patients with cardiac cachexia. In this pilot study, 19 patients with >7.5% weight loss received six weeks of therapy (protein 20 g, carbohydrate 72 g, fat 26 g/day) and six patients received placebo. The patients in the treatment group gained weight, and their cholesterol increased and cytokines decreased.
Modulation of the Th1-Th2 balance depends on therapy given in just the right dose. Anker proposed a self-adjusting treatment system using apoptotic cells. Apoptotic cells have receptors recognized by macrophages and dendritic cells, which trigger signals that change the pool of naïve T cells away from a Th1 response toward a Th2 response. This switch from Th1 to Th2 response only occurs in patients whose Th1-Th2 response is out of balance.
A device (Celacade) currently in clinical testing triggers controlled apoptosis in a small amount of patient blood, which is then injected intramuscularly into the patient. A randomized phase II study showed a significant reduction in mortality and hospitalization (Figure 1) in treated patients. A large multinational phase III trial of this treatment (ACCLAIM) has recently ended and will be reported in September 2006 at the World Congress of Cardiology (Figure 2).
|
|
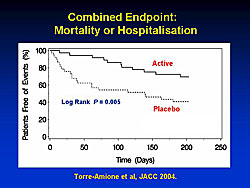

| Footnote: Figure 1: Reprinted from JACC 2004; 44(6):1181-1186. Effects of a novel immune modulation therapy in patients with advanced chronic heart failure: results of a randomized, controlled, phase II trial. Torre-Amione G, Sestier F, Radovancevic B, Young J. With permission from American College of Cardiology Foundation. |
|
|
|
Copyright © 2006 Japanese Circulation Society All Rights Reserved. webmaster@j-circ.or.jp |