Triggers and mediators of preconditioning
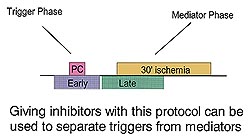
Fig 3 reveals that the trigger and
mediator functions of preconditioning can be separated
by giving inhibitors either early to bracket the
preconditioning ischemia or late to load the heart
with inhibitor just before the long ischemic period.
Giving an adenosine receptor antagonist using the
early protocol completely blocks protection, showing
that adenosine works as a trigger. On the other
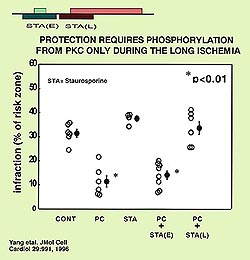
hand staurosporine, a PKC blocker will only abort
preconditioning if given in the late protocol indicating
that PKC is a mediator (see Fig 4). The question
is; does opening the mKATP channels act as a trigger
or a mediator of protection? Before we address that
question we need some more background information
on preconditioning's pathway.
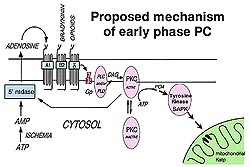
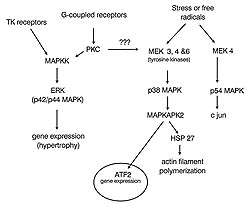
Location of tyrosine kinase
Studies by Baines in my laboratory
showed that preconditioning involves a tyrosine
kinase. Genistein a tyrosine kinase inhibitor only
blocked protection from preconditioning if the late
protocol was used indicating that the tyrosine kinase
acts as a mediator like PKC. We had reason to believe
that the location of the tyrosine kinase in question
was at p38 MAPK. Activation is by an upstream kinase
called a MEK (see Fig 5) which phosphorylates p38
MAPK on its tyrosine 182 and threonine 180. The
MAPKs are thought to be turned on only in a preconditioned
heart and will have both immediate protective effects
as well as to cause gene expression; presumably
important in creating the second window by causing
a protective protein to be produced that protects
the heart long-term.
In an isolated rabbit heart model
we showed there was no activation of p38 MAPK during
ischemia in the non-preconditioned heart. When the
heart is preconditioned there was no significant
activation of p38 MAPK just prior to ischemia. But,
during ischemia, the so-called mediator phase, a
3-fold increase in p38 MAPK activity was seen at
10 minutes and 20 minutes. If protection was blocked
with an adenosine receptor blocker so that the preconditioned
hearts were not actually protected, the increase
in phosphorylation was lost. Thus, an increase in
p38 MAPK activation during ischemia is associated
with protection.
We wanted to measure an index of p38
MAPK's kinase activity. To accomplish this we developed
an assay for the downstream kinase, MAPKAPK2 (mitogen
activated protein kinase activated protein kinase
2). Tissue homogenate was separated by liquid chromatography
into serial fraction using a Mono-S column. The
ability of each fraction to phosphorylate a substrate
peptide specific for MAPKAPK2 was then measured.
At either baseline or after 20 minutes of ischemia
there is no appreciable activity in any fraction
from non-preconditioned hearts. However, there were
two very prominent peaks of MAPKAPK2 activity in
the preconditioned hearts after 20 minutes of ischemia.
Western blot analyses of the fractions against the
two known isoforms of MAPKAPK2 show they are both
present in the first of the two activity peaks.
Good evidence again that p32 MAPK is being turned
on during ischemia in the preconditioned heart and
that it is acting as a mediator. Work by Yellon
looking at SB203580, a p38 MAPK blocker, shows that
giving it with the early protocol has no effect
against preconditioning's protection in the rat
heart, but giving it late completely blocks the
preconditioning protection. Again confirming this
mediator role.
Timing of KATP channel opening
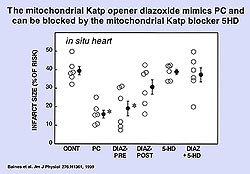
To determine when the KATP channel
must be open for protection, we performed a similar
study to the experiment in figure 3 using the mKATP
blocker 5HD. Isolated rabbit hearts were used. A
5-minute diazoxide infusion was followed by a 10-minute
wash out and then 5-HD was given either early or
late. The early protocol (bracketing the diazoxide
infusion) completely blocked protection, but when
5HD was given late there was no effect on protection.
It appears that the KATP channel does not need to
be open during the 30-minute period of ischemia.
Clearly mKATP opening from diazoxide is acting as
a trigger.
We repeated the experiment using ischemic
preconditioning (5 minutes of coronary occlusion
plus 10 minutes of reperfusion) in isolated rabbit
hearts. 5-HD when given early completely blocked
preconditioning's protection. But, when 5-HD was
given late there was no effect on protection. The
mKATP channel opening is clearly acting as a trigger
with ischemic preconditioning as well as with diazoxide
treatment.
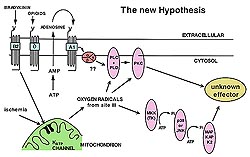
Is mKATP channel opening a signal
transduction step?
If mKATP channel opening acts as a
signal transduction step, diazoxide treatment would
be expected to activate the downstream kinases.
A study was done to determine whether a kinase blocker
would block protection from diazoxide. When diazoxide
was given with chelerythrine, a PKC blocker, to
isolated hearts, neither the early nor the late
protocol had any effect on protection. But, a tyrosine
kinase blocker, genistein, completely blocked protection
from diazoxide when given with the late protocol.
Clearly diazoxide causes kinase-dependent protection,
indicating that the mKATP channel is upstream from
at least one tyrosine kinase. Opening the mKATP
channel triggers protection, which causes activation
of the kinases during the index ischemia.
Free radicals
In perhaps the most important experiment,
diazoxide was shown to protect in a free radical-dependent
manner. N-2-mercaptopropionylglycine (MPG), an intracellular
free radical scavenger, was given 5 minutes prior
to diazoxide and continued until halfway through
the ischemic period. MPG completely blocked protection.
A recent paper by Yao probably explains this effect.
Dichlorofluorescein, a dye that becomes fluorescent
when it reacts with free radicals, was used as an
index of free radical production. In the cardiomyocyte
acetylcholine occupies a Gi-coupled receptor (M2)
that is very similar to the adenosine A1 receptor,
and because it activates PKC, acetylcholine can
mimic preconditioning. After neonatal chicken myocytes
were given acetylcholine, there was a burst of free
radicals. When myxothiazol, an electron transport
blocker, was given the production of free radicals
from acetylcholine was blocked. This indicates that
free radicals were coming from the mitochondria.
When MPG was given, the free radicals are soaked
up and the signal removed. Most importantly, when
the mKATP channel was blocked by 5-HD the burst
of free radicals was lost. This indicates that giving
a receptor agonist causes opening of mKATP channels
which causes a burst of free radicals.